
1. Introduction
Cervical cancer is the second most common type of cancer in Indonesia. Based on data from WHO, there were 36,633 new cases in 2020, while the death rate from cervical cancer in the same year reached 21,003 deaths [1]. The main cause of cervical cancer is the High-Risk Human Papillomavirus (HPV) infection, identified in over 99,7% of cervical cancers [2]. HPV 16 is the most prevalent type which is detected in 60.5% of cases [3]. Although effective prophylactic HPV vaccines are readily available commercially such as Merck's Gardasil (quadrivalent vaccine) and GlaxoSmithKline's Cervarix (bivalent vaccine), the high cost of vaccination is a major barrier. Besides, this vaccine is only effective if it is given to uninfected individuals [4]. Therefore, an effective and affordable therapeutic approach is urgently needed.
The ideal anticancer drugs are those that are specific and cytotoxic only in cancer cells [5]. In cervical cancer, there are two main oncoproteins in HPV (i.e., E6 and E7), which are only expressed in infected cells that could be potential therapeutic targets. Both proteins cause cell immortalization and are continuously expressed during cancer development [6].
E6 is an 18 kD protein consist of approximately 150 amino acids [7]. The main target of E6 is tumor suppressor protein p53. E6 HPV 16 causes ubiquitination and degradation of p53 in two ways. First by binding directly to p53 (E6AP independent) or second by forming the E6 / E6AP / p53 complex (E6AP dependent). The degradation of p53 causes the uncontrolled proliferation of cells that are resistant to apoptosis [8]
E7 is the first oncoprotein discovered. This protein is relatively small, about 100 amino acids, divided into three conserved regions: CR1, CR2, and CR3 [8]. Half of the
N-terminal E7 terminals (amino acids 1-40) constituting the CR1 and CR2 regions are naturally unfolded (distinctly disordered) and are flexible with few conformational transitions [9]. Meanwhile, CR3 E7 HPV plays a role in the dimerization of E7 and becomes a medium for direct interaction with several proteins. In particular, CR3 interacts with the C-terminal portion of pRb. E7 full-length protein is 100 times more potential to bind pRb than E7 which only contains CR1 / CR2, this indicates an important role for CR3 in interactions with pRb [10]. E7 binds to pRb then causes ubiquitination of pRb so that E2F is released from pRb and becomes active. E2F is a transcription factor of genes that cells need to enter the synthesis phase in the cell cycle. The synthesis phase prompts the cell to activate DNA replication and initiates the process of self-division [8]. The combination action of E6 and E7 in cervical cells leads to cell transformation and cancer development. Hijacking E6 and E7 binding to cellular protein targets by an inhibitor could have an impact on their oncogenic activity [11].
Various types of therapies that target these two proteins have been developed, e.g., therapeutic vaccines targeting HPV 16/18 E6 / E7 for the treatment of advanced cervical cancer, genome editing using antisense oligonucleotides, ribozymes, DNAzymes, siRNA (small interfering RNA), shRNA (short-hairpin RNA), immunotherapy with synthetic E6 / E7 HPV16 / 18 DNA sequences, and tumor-infiltrating T cells (reviewed by Pal and Kundu [8]). Therapy with the E6 / E7 target gives promising results, but the high cost will be an obstacle to its application. Hence, discovering effective and more affordable alternatives becomes crucial.
The use of natural products promises a comparatively safer alternative therapeutic approach to cervical cancer. This therapy also offers less complicated treatments using abundantly available and inexpensive medicine compared to genome editing technologies or immunotherapeutic methods [8]. Natural compounds, such as plant extracts in either pure form or standardized extracts, provide unlimited opportunities for new drug discovery due to the unrivaled availability of chemical diversity [12]. The chemical diversity of natural compounds can be an excellent source of novel scaffolds. These chemical scaffolds are optimized further to get the new drug candidates.
Indonesia is a country with extraordinary high biodiversity. Indonesia's tropical rainforests, covering an area of approximately 143 million hectares, are home to about 80% of the world's medicinal plants [13]. Active compounds from Indonesian medicinal plants could be potential E6 and E7 inhibitors. A computational experiment or in silico method is conducted as a shortcut to seeking this great potential by creating computational models or simulations that can be used to make predictions, suggest hypotheses [14]. This method is more effective and efficient for screening new drugs for pre-eliminary research, the range of compounds being tested can be wider with a shorter time and lower cost. In this study, we explored Indonesian medicinal plants active compounds potency an oncoprotein E6 and E7 HPV 16 inhibitors by molecular docking.
2. Material and Methods
2.1. Hardware
DELL laptop with a specification of processor processor Intel® Core™ i5-7200U CPU @ 2,5 GHz RAM 8GB and 64-bit Windows 10 operating system was used in this research.
2.2. Software
Bioinformatics programs used included Marvin Sketch (ChemAxon, Budapest, Hungary), Modeller 9.16 (University of California San Francisco, USA), BIOVIA Discovery Studio Visualizer 2020 (Dassault Systèmes, San Diego), Autodock Tools1.5.6 (The Scripps Research Institute, USA), Autodock Vina (TheScripps Research Institute USA) [15], LigPlot+ 2.2 (EMBL-EBI, UK) [16], and Chimera 1.14 to visualize the ligand position on the protein [17].
2.3. Protein preparation
E6 HPV 16 3D structure (chain F) was separated from E6/E6AP/p53 complex crystal structure retrieved from Protein Data Bank (PDB), ID 4XR8, and a resolution of 2,25 Å. Visualization and separation were processed by BIOVIA Discovery Studio Visualizer 2020. Polar hydrogen atoms were added to the structure and convert to *.pdbqt format by Autodock Tools 1.5.6.
E7 protein used in this research was only the CR3 domain. E7 HPV 16 sequence was harvested from the Uniprot server. The template used for modeling the 3D structure was ID2B9D (PDB id), a crystal structure of E7-CR3 domain of HPV 1A dimer with 37% identity to E7-CR3 domain of HPV 16. E7-CR3 domain of HPV 16 3D dimer structure was built by MODELLER 9.25. YASARA Energy Minimization server was used for protein model structural refinement and energy minimization [18]. The refined structure then confirmed for reliability by ProSA-Web [19] and plot to The Ramachandran plot (Zhiping weng's laboratory, USA) [20].
2.4. Ligand preparation
There were 711 active compounds of 187 Indonesian medicinal plant species that were collected and filtered by molecular weight, solubility, GI absorption, and druglikeness by Lipinski's Rules of Five. The selection was carried out by submitting the 2D structure of the active compound to SwissADME webserver [21]. Compounds that have MW of more than 200 g/mol, moderate to high
solubility, high GI absorption, and fulfill drug-likeness criteria were selected and prepared for docking. The 3D conformations were drawn on Marvin Sketch by employing the dreiding force field as minimization energy.
2.5. Binding Site Identification
Identification of residues involved in E6-E6AP and E6 p53 interaction was based on E6/E6AP/p53 crystal structure complex resolved by Martinez-Zapien et al. [22] which also determined hot spot residues by mutational study and binding assay. CR3 E7 HPV 1A interaction with pRb was identified by Liu et al. [10]. Residues involved in CR3 E7 HPV 16-pRb interaction determined by amino acid sequence
alignment between CR3 E7 HPV 16 and CR3 E7 HPV 1A. Pocket searching in the binding site areas of E6-E6AP, E6 p53, and CR3 E7 HPV 16-pRb were performed by fpocket webserver [23]. Those pockets were then used for the docking study.
2.6. Molecular Docking Simulation
Molecular docking was executed by Autodock Vina, a fast and accurate docking tool [15]. Grid box parameters were set in specific searching area to encompass binding site for effective docking simulation (Table 1). The docking results were visualized by LigPlot+ 2.2 in 2D and Chimera 1.14 in 3D molecular structures.
Table 1. Grid Box Parameters
| No | Receptor | Protein Binding Patner (Binding Site) | Grid Center Coordinate | Grid Box Size (Å) | ||||
|---|---|---|---|---|---|---|---|---|
| x | y | z | x | y | z | |||
| 1. | E6 | E6AP | - | -7,059 | -12,871 | 20 | 20 | 20 |
| 52,065 | ||||||||
| 2. | E6 | p53 | - | 1,947 | -17,234 | 20 | 20 | 20 |
| 61,861 | ||||||||
| 3. | CR3 E7 | pRb | 22,31 | 24,86 | 50,682 | 20 | 20 | 20 |
3. Results and discussion
The major roles of E6 and E7 oncoprotein in cervical carcinogenesis make these proteins a potential target for cervical cancer therapy [24]. Blocking both protein interactions with its cellular protein targets will be expected to have a significant impact to suppress cancer development and lead to senescence. This research aims to find Indonesian medicinal plants' active compounds that have good potency to inhibit the interaction of oncoprotein E6 to E6AP, E6 to p53, and oncoprotein E7 to pRb. In this research, the inhibition potency of the active compounds was evaluated by molecular docking to E6 and E7.
3.1. Active compounds selection
Active compounds were filtered by molecular weight, solubility, GI absorption, and drug-likeness (Lipinski's rules of five). Low molecular weight compounds bound weakly to protein [25], so only compounds that have a molecular weight above 200 g/mol used in this study. Compound solubility in water is important for drug formulation to achieve the desired concentration of drug in systemic circulation for desired pharmacological response [26]. The GI absorption parameter was observed in this study because this parameter is related to the absorption rate of the compound in the intestine and its bioavailability. High GI absorption is very important for oral drug candidates. The oral route is the most common and preferred way of administration over other routes because of its many advantages. These advantages include safety, ease of administration, high patient compliance, painlessness, costeffectiveness, and flexibility in dosage form design [27]. The Lipinski's rule of five helps to distinguish a drug-like compound from a non-drug based on five rules: molecular mass < 500 Da, lipophilicity (LogP < 5), hydrogen bond donors < 5, hydrogen bond acceptors < 10, molar refractivity should be within 40 to 130.28 [28]. Active compounds selection results in 164 compounds that meet the criteria.
3.2. Binding Affinity and Molecular Interaction Profile
E6 oncoprotein was docked in two binding sites: E6-E6AP interface and E6-p53 interface. Residues involved in E6- E6AP interaction are R10, K11, V31, Y32, L50, C51, V53, R55, V62, L67, Y70, S74, R77, H78, L100, R102, Q107, R129, and R131. L50, R102, and R131 are considered hot spot residues because of their high contribution for E6AP binding, mutation of these residues leads to binding perturbation, prevents p53 degradation, and disrupts the oncogenic activity of E6 [29].
Docking results of active compounds to E6 in E6AP interface are shown in Table 2. Table 2 shows active compounds with the best docking score to E6 at E6AP binding site and a standard anticancer drug (i.e., jaceosidin) as a positive control. Jaceosidin is a flavonoid from
Artemisia princeps which experimentally through the ELISA method has the potential as an inhibitor of oncoproteins E6 and E7. In vitro testing on SiHa and CaSki cell lines containing the HPV 16 genome shows jaceosidin inhibits cancer formation [30]. Nine compounds, i.e., elephantin, roemerine, ginkgolide A, phaseollin, anonaine, chitranone, elephantopin, tetrahydroalstonine and vindolinine have significantly lower docking score (-8,3 to -8,7 kcal/mol) than positive control (-7,2 kcal/mol). The lower docking score or binding affinity indicate the more stable and favorable the interaction. The nine compounds have better affinity to E6 than jaceosidin because they have more hydrophobic interactions. Figure 2.a shows that the elephantin interact with nine hydrophobic residues of E6 (Y32, L50, V53, V62,
L67, Y70, S71, S74, R102) or 47,37% compared to total 19 residues involved in E6-E6AP protein-protein interaction. While jaceosidin has only three hydrophobic interactions (L50, Q107, R131) or 31,58% (Figure 1.a). Elephantin atomic groups are mostly in the hydrophobic E6 protein residue (dark orange color), shown in Figure 2.b. Hydrophobic interaction can increase the affinity in the protein-ligand complex. The addition of the number of hydrophobic atoms at the active site of the target and drug interface affects the biological activity of the drug candidate. The increase in binding affinity to the target complex and the drug that results from the optimization of hydrophobic interactions shows better drug efficacy [31]
Table 2. Docking Results of Indonesian Medicinal Plant Active Compounds against Oncoprotein E6 on the Binding Site of E6AP
| Active Compound | Plant Spesies | Docking Score/Binding Affinity (kcal/mol) | Hydrogen Bonds | Hydrophobic Interaction | Total Interaction |
|---|---|---|---|---|---|
| Jaceosidin (positive control) | Artemisia princeps (Mugwort Korea) | -7.2 | L100, W132, R102, C51 | L50, Q107, R131 | 31.58% |
| Elephantin | Elephantopus scaber (tapak liman) | -8,7 | C51, Q107, R131 | Y32, L50, V53, V62, L67, Y70, S71, S74, R102 | 47.37% |
| Roemerine | Nelumbo nucifera (teratai) | -8,7 | - | V31, Y32, F45, L50, C51, V53, V62, Y70, S71, Q107, R102 | 47.37% |
| Ginkgolide A | Ginkgo biloba (ginkgo biloba) | -8,6 | - | Y32, L50, C51, L67, Y70, S71, S74, Q107, R131 | 42.10% |
| Phaseollin | Erythrina fusca (cangkring) | -8,6 | - | V31, Y32, F45, L50, C51, V53, A61, V62, L67, Q107, R102, R131 | 52.63% |
| Anonaine | Annona squamosa (srikaya) | -8,5 | - | V31, Y32, L50, C51, V53, V62, L67, S71, R102, Q107 | 47.37% |
| Chitranone | Plumbago zeylanica (daun encok) | -8,4 | C51, R102 | L50, V62, L67, L100, Q107, R 131 | 42.10% |
| Elephantopin | Elephantopus scaber (tapak liman) | -8,4 | C51, R131 | Y32, L50, V53, V62, L67, Y70, S71, S74, Q107 | 52.63% |
| Tetrahydroalstoni ne | Catharanthus roseus (tapak dara) | -8,3 | R102 | V31, Y32, F45, L50, C51, V53, A61, V62, L67, S71, S74, Q107, | 57.89% |
| Vindolinine | Catharanthus roseus (tapak dara) | -8,3 | Q107 | R131 Y32, F45, L50, C51, V62, L67, Y70, S71 | 31.57% |
.
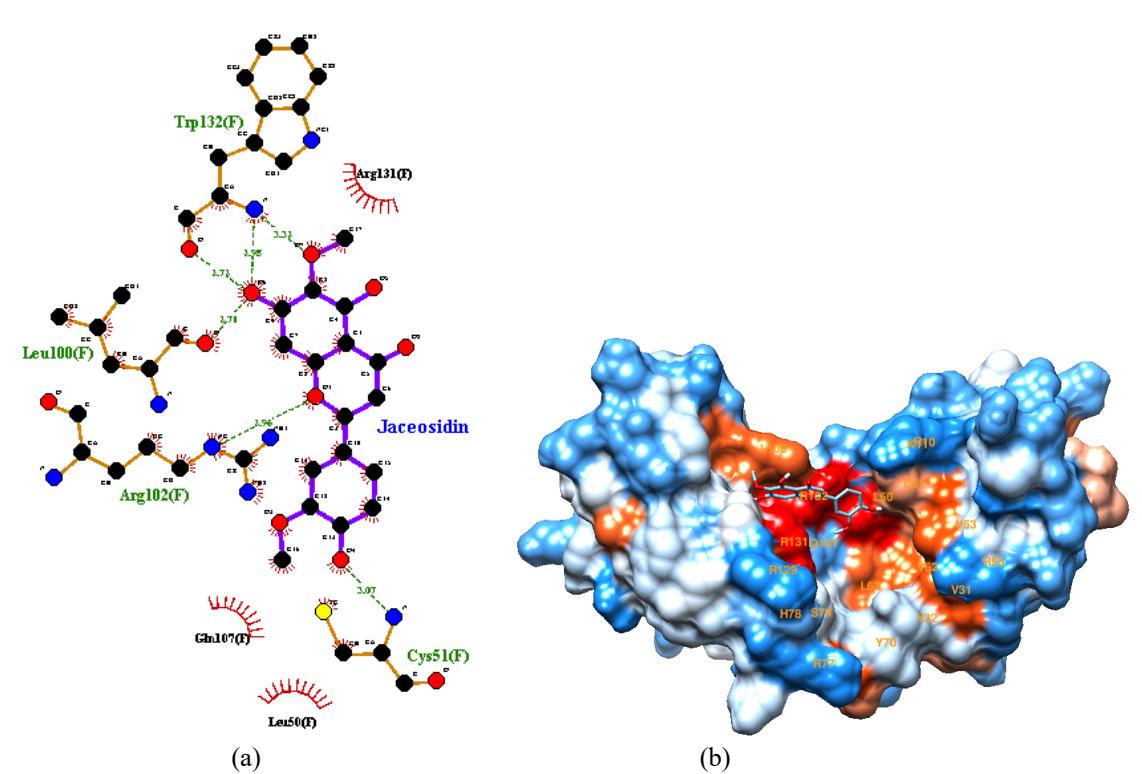
Figure 1. 2D visualization (a) and 3D visualization (b) of jaceosidin-E6 interaction on E6AP binding site.
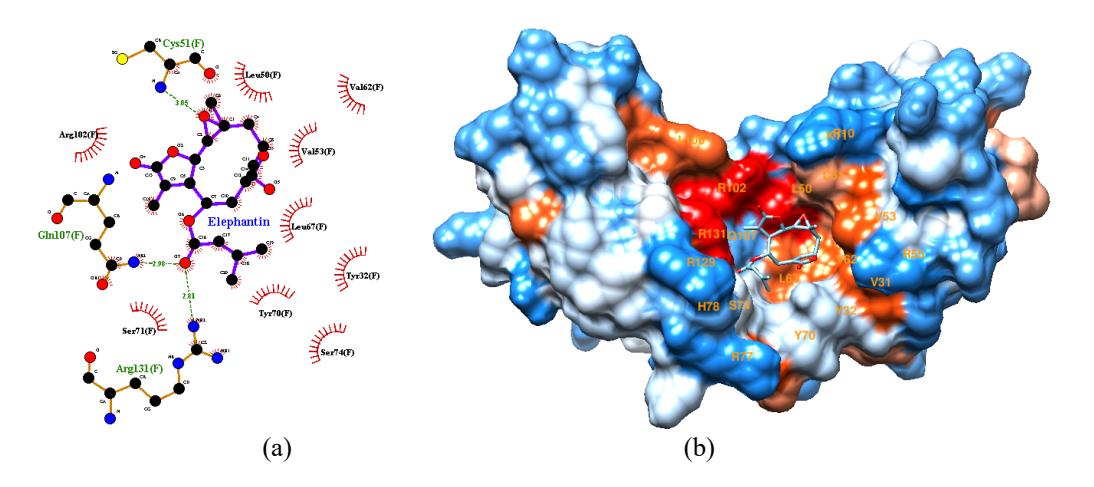
Figure 2. 2D visualization (a) and 3D visualization (b) of elephantin-E6 interaction on E6AP binding site.
Having three hydrogen bonds, elephantin has better specificity among the nine compounds. Hydrogen bonding is a fundamental interaction in protein-ligand complexes and is the main reason for the selectivity of ligand proteins, due to its specific nature, short spacing, and directionality [32-33]. Docking programs generally model hydrogen bond interactions better than hydrophobic interactions. This relates to the need for specificity in drug design [33].
The compound's interaction with hot spot protein residues also affects its potential as inhibitor. Hot spot residues are crucial residues for protein-protein interactions, so that intervention in these residues has an impact on their binding. L50, R102, and R131 are hot spot residues in E6-E6AP interaction. Elephantin, phaseolin, chitranone, and tetrahydroalstonine have interactions with all these residues via hydrogen bonding and/or hydrophobic interaction so that these compounds could have better potency as E6 inhibitors.
Molecular docking was also executed on p53 binding site, to simulate inhibition of E6-p53 direct interaction by active compounds. Molecular interaction profile of best docking
score compounds shown in Table 3. D44, F47, and D49 were known to be E6 hot spot residues in E6-p53 interaction base on research conduct by Martinez-Zapien et al. [22]. E6 also interacts with p53 via I23, H24, and Y43. All these six residues lay on the center of E6-p53 interaction. Table 2 exhibits six compounds that are chitranone, sesamin, 16acetylgitoxigenin, tetrahydroalstonine, methylophiogonone A, and phaseollin have a significantly better binding affinity (-6,9 kcal/mol to -7,3 kcal/mol) to E6 on p53 binding site than the positive control. Chitranone, sesamin, and 16-Acetylgitoxigenin have lower docking scores despite their fewer interactions that control positive, probably because of the better fitness compound to E6 surface interaction or shorter inter-atomic surface distance between compounds and protein. Surface distance is one of the parameters calculated in Autodock Vina scoring function. Binding
affinity and hydrophobic interaction of the six best compounds do not differ significantly, but tetrahydroalstonine, methylophiogonone A, and phaseolline show better specificity by having more hydrogen bonds.
E7 key role in cancer initiation work in concert with E6. Blocking both protein interaction with cellular protein is important to completely suppress their oncogenic activities. The CR3 E7 residues that interact with pRb are R66, L67, I76, R77, E80, D81, M84. Based on the mutational study conducted by Liu et al. [10], a single mutation in the R66 residue and a mutation in the E80 / D81 residue had a significant effect in inhibiting the interaction of E7 with pRb. Therefore R66, E80, D81 are defined as hot spot residues. Molecular docking performed to CR3 E7 HPV16 results 11 best compounds shown in Table 4.
Table 3. Docking Results of Indonesian Medicinal Plant Active Compounds against Oncoprotein E6 on the Binding Site of p53
| Active Compound | Plant Spesies | Docking Score/Binding Affinity (kcal/mol) | Hydrogen Bonds | Hydrophobic Interaction | Total Interaction |
|---|---|---|---|---|---|
| Jaceosidin (positive control) | Artemisia princeps (Mugwort Korea) | -6,1 | D44, R48, C106, S111 | L108, E114, M137, S138 | 16,67% |
| Chitranone | Plumbago zeylanica (daun encok) | -7,3 | K108 | Y43, F47, S111, E114, M137 | 33,33% |
| Sesamin | Piper retrofractum (cabe jawa) | -7,1 | R48 | Y43, D44, F47, S138 | 50% |
| 16-Acetylgitoxigenin | Nerium indicum (jure) | -7,0 | R48 | R40, Y43, D44, F47, C106, K108 | 50% |
| Tetrahydroalstonine | Catharanthus roseus (tapak dara) | -7,0 | S138, R48, D44 | R40, Y43, F47, C106, K108 | 50% |
| Methylophiogonone A | Ophiopogon japonicus (ophiopogon) | -6,9 | R48, K108, S138 | D44, F47, C106, S111, E144, M137 | 33,33% |
| Phaseollin | Erythrina fusca (cangkring) | -6,9 | E114, M137, S138 | Y43, D44, F47, K108, C136 | 50% |
Table 4. Docking Results of Indonesian Medicinal Plant Active Compounds against Oncoprotein E7 on the Binding Site of pRb
| Active Compound | Plant Spesies | Docking Score/Binding Affinity (kcal/mol) | Hydrogen Bonds | Hydrophobic Interaction | Total Interaction |
|---|---|---|---|---|---|
| Jaceosidin (positive control) | Artemisia princeps (Mugwort Korea) | -5,8 | Q70, D81 | R66, V74, T78, L82, L87, T86, G88 | 28,57% |
| Columbin | Tinospora crispa (brotowali) | -6,9 | R66 | N53, V55, Q70, T78, D81, L82, T86, G88 | 28,57% |
| Methylophiogonone A | Ophiopogon japonicus (ophiopogon) | -6,9 | D81 | N53, V55, R66, T64, Q70, V74, R77, T78, L82, T86, G88 | 42,86% |
| Serpentine | Rauvolfia serpetina (pule pandak) | -6,8 | - | N53, V55, R66, V74, R77, T78, D81, L82, T86, L87, G88 | 42,86% |
| Uzarigenin | Nerium indicum (jure) | -6,8 | N53 | R66, V74, T78, D81, L82, T86, L87, G88 | 28,57% |
| Apigenin | Apium graveolens (seledri) | -6,6 | N53, D81 | V55, T64, L65, R66, T78, L82, T86, G88 | 28,57% |
| Coumestrol | Taraxacum officinale (jombang) | -6,6 | N53 | V55, T64, R66, T78, L82, T86, L87, G88 | 14,29% |
| Kaempherol | Garcinia latissima (dolo magota) | -6,6 | N53, T64, D81 | V55, L65, R66, T78, L82, T86, G88 | 28,57% |
| Phaseollin | Erythrina fusca (cangkring) | -6,6 | N53 | V55, R66, T78, D81, L82, L87, G88 | 28,57% |
| Pelargonidin | Phaseolus vulgaris (buncis), Impatiens balsamina (pacar air) | -6,5 | N53, T64, D81 | V55, L65, R66, T78, L82, T86, G88 | 28,57% |
| Rhamnocitrin | Alpinia galanga (lengkuas) | -6,5 | N53 | V55, T64, L65, R66, T78, D81, L82, T86, G88 | 28,57% |
| Sesamin | Piper retrofractum (cabe jawa) | -6,5 | - | R66, V74, R77, T78, D81, L82, T86, G88 | 42,86% |
Table 4 shows the 11 compounds with the best docking scores (-6.5 to -6.9 kcal/mol). This score was significantly better than the positive control docking score. Kaempherol and pelargonidin have relatively better specificity compared to positive control and other compounds because of their hydrogen bonds. All compounds have interactions with hot spot residues either through hydrogen bonds or hydrophobic interactions, so that they have good potency as E7 inhibitors. The total interactions that occurred between the compounds ranged from 28.57% to 42.86%, except for coumestrol which was slightly smaller, namely 14.29%. Based on the results in Table 4, it is concluded that the compounds columbin, methylopiogonone A, serpentine, uzarigenin, apigenin, coumestrol, kaempherol, phaseollin, pelargonidin, rhamnocitrin, and sesamin are potential inhibitors of E7.
Among the compounds that have the potential to act as inhibitors of E6-E6AP, E6-p53, and E7-pRb, phaseollin can be potential inhibitors of the three interactions. Meanwhile, compounds that have the potential to act as inhibitors of E6-E6AP and E6-p53 are chitranone and tetrahydroalstonine.
Sesamin and methylophiogonone A have the potential to inhibit E6-p53 and E7-pRb interactions. Further research is needed to determine the potential of the best compounds obtained in this study in vitro and in vivo.
4. Conclusion
The diversity of Indonesian medicinal plants provides as abundant active compounds source for anti-cancer drug discovery. Cervical cancer caused by HPV 16 infections, develop as impact of continuous expression of E6 and E7 oncoproteins in infected cells. In this study, we explored Indonesian medicinal plants active compounds potency an oncoprotein E6 and E7 HPV 16 inhibitors by molecular docking. By observing the docking score, hydrogen bonding, hydrophobic interactions, and interactions of compounds with E6/E7 hot spot protein residues, we suggest potential E6/E7 inhibitors. Elephantin, roemerine, ginkgolide A, phaseollin, anonaine, chitranone, elephantopin, tetrahydroalstonine and vindolinine were found to be potential as E6 inhibitors on E6AP binding site, while chitranone, sesamin, 16-acetylgitoxigenin, tetrahydroalstonine, methylophiogonone A, and phaseollin exhibit potency as E6 inhibitors on p53 binding site. Columbin, methylopiogonone A, serpentine, uzarigenin, apigenin, coumestrol, kaempherol, phaseollin, pelargonidin, rhamnocitrin, and sesamin showed potency as E7 inhibitors. Phaseollin was found to be potential as E6 and E7 inhibitors.