
Introduction
Methane (CH<sub>4</sub>) is a potent greenhouse gas and a major environmental contaminant. The global oil and natural gas industry is a significant contributor to CH<sub>4</sub> emissions, releasing gas through leakage, incomplete combustion, flaring, and venting in various processes (Roshchanka & Evans, 2014). Environmental pollution is exacerbated by direct emissions of this gas, which exhibits a greenhouse effect 20–30 fold greater than that of carbon dioxide (Lu, 2021). CH<sub>4</sub> emissions from sources such as landfills can lead to groundwater and air pollution, which can negatively impact climate and pose health hazards (Aljaradin, 2012). Although CH<sub>4</sub> is not directly toxic to humans, it is frequently co-emitted with other pollutants that can negatively affect adjacent communities, thus underscoring environmental justice concerns (Casey et al., 2021). Addressing systematic breaches in gas compressor units and pipelines is essential for reducing CH<sub>4</sub> emissions and their impact on the environment and nature (Strizhenok & Korelskiy, 2019). The conversion of CH<sub>4</sub> to hydrogen (H<sub>2</sub>) molecules without the formation of greenhouse gases is highly advantageous, particularly because natural gas is predominantly composed of CH<sub>4</sub>. By minimizing the overall generation of greenhouse gases, this process addresses environmental concerns and provides an alternative energy source (Kikuchi, 2002).
Copyright ©2025 Published by IRCS - ITB ISSN: 2337-5779
Several studies have emphasized the potential of catalytic methane decomposition (CMD) as a viable approach for the production of H2 along with solid carbon or carbon nanostructures (Gamal et al., 2021; Harbin et al., 2023; Shelepova et al., 2022; Sun, 2024). This process is environmentally friendly and produces COx-free H2 and solid carbon products (Dipu, 2021; Harbin et al., 2023). These materials have various applications in catalysis, electronics, and fuel cells (Gamal et al. 2021). CH4 decomposition is widely acknowledged as a reliable method for producing H2, offering a simpler and potentially more cost-effective alternative to steam reforming, as highlighted in recent studies (Hasnan et al., 2020; Rad et al., 2022). Furthermore, researchers have found that the thermal decomposition of CH4 is promising for large-scale production of H2. This process can mitigate the release of greenhouse gases, as noted by Sanchez‐Bastardo et al. (2021) and Santos et al. (2022) and Hasnan et al. (2023). Researchers have investigated the use of different catalysts, including Cr–O–Ni and Ni-based perovskites, to enhance the production of H2 and carbon nanotubes during CH4 decomposition (Harbin et al., 2023; Sun, 2024). In addition, researchers have studied the catalytic decomposition of CH4 under various conditions, such as the production of H2 using nanomaterials (Fujimoto & Ohba, 2022). Arifin et al. examined the processes of CH4 dehydrogenation and H2 formation on Pt(100) (Arifin & Darminto, 2023) and Pt7-Ni(110) (Arifin et al., 2024) surfaces, emphasizing the significance of metal surfaces in promoting the separation of hydrogen atoms from CH4 molecules (Arifin et al., 2024; Arifin & Darminto, 2023). In addition, Arifin et al. (2015) performed first-principles calculations to gain insight into the process of CH4 decomposition on a Ni(111) surface, providing valuable information on the fundamental aspects of CH4 decomposition on metal catalysts. The above-mentioned studies highlight the potential of CH4 decomposition as a sustainable and effective method for H2 production. In the field of computational catalysis, numerous studies have utilized density functional theory (DFT) to investigate reaction mechanisms and catalyst characteristics. For instance, DFT has been applied to elucidate persulfate-based advanced oxidation processes and mechanisms of organic compound isomerization (Gallego-Villada et al., 2024; Zhang et al., 2021).
Nickel plays a vital role as a catalyst in CH4 decomposition and H2 formation owing to its exceptional catalytic properties. Furthermore, numerous studies have emphasized the importance of nickel-based catalysts in facilitating CH4 decomposition reactions for H2 production. For example, Chen and Lua employed electroless plating to create nickel catalysts for H2 generation via CH4 decomposition (Chen & Lua, 2020). In a recent study, Phichairatanaphong et al. successfully created a catalyst using nickel-supported mesoporous silica–aluminosilicate to improve H2 production through CH4 decomposition (Phichairatanaphong et al., 2021). Recently, Yan et al. examined the process of generating COx-free H2 and graphene nanoplatelets via CH4 decomposition using Ni-lignin-derived nanoparticles, placing particular emphasis on the role of nickel in the CH4 decomposition process (Yan et al. 2022). In addition, previous studies have indicated that the addition of metals such as copper and iron to nickel-based catalysts can improve their catalytic activity in CH4 decomposition reactions. Recently, Hasnan et al. prepared bimetallic nickel catalysts supported on mesostructured silica nanoparticles with copper and iron, which exhibited enhanced activity and stability for CH4 decomposition (Hasnan et al., 2023). In another recent study, Vlaskin emphasized the effectiveness of using a Ni catalyst to enhance the efficiency of CH4 decomposition by reducing the required temperature (Vlaskin, 2023). In addition, the size and dispersion of nickel particles are important factors in catalytic reactions. Recently, Liang et al. found that the deactivation rate of nickel catalysts in CH4 decomposition was influenced by the size of nickel particles (Liang et al., 2020). Further, Wang et al. examined a pre-coking strategy that demonstrated the significance of nickel in catalytic processes; their findings highlighted how this strategy can improve the stability performance of supported nickel catalysts in hydrogenation reactions (Wang et al., 2021).
Extensive research has been conducted on metallic nickel catalysts, owing to their well-established high catalytic activity for CH4 decomposition (Wang & Lua, 2012). However, recent studies have questioned the involvement of Ni clusters in this process (Harrath et al., 2023). Thus, further investigation is necessary to elucidate the specific mechanisms through which nickel clusters facilitate CH4 decomposition and to provide insights into the fundamental processes at the nanoscale level. This study aimed to examine the molecular mechanisms and kinetics involved in the catalytic conversion of CH4 over nickel catalysts. The objective of this study is to elucidate the dynamic mechanism of nickel clusters in the conversion of CH4 to various products, including H2, using molecular dynamics (MD) simulations. Thus, the current investigation aimed to clarify the precise interactions, dynamics, and energetics of CH4 molecules in the presence of Ni surfaces, particularly in the context of reforming processes.
Methods
In this study, we used MD simulations to explore the reaction mechanisms of CH<sub>4</sub> on nickel clusters. Using the ReaxFF reactive force field, chemical reactions were dynamically modeled by adjusting the bond orders throughout the simulations (van Duin et al., 2001). This capability makes ReaxFF notably well suited for studying systems where chemical reactions are critical, such as those involving catalysis and combustion (van Duin et al., 2001). In this study, we optimized the ReaxFF potential parameters to accurately capture the interactions between CH<sub>4</sub> molecules and nickel clusters in hydrocarbon–metal systems (Mueller et al., 2010).
The simulation system included nickel clusters of various sizes (Ni<sub>37</sub>, Ni<sub>55</sub>, and Ni<sub>80</sub>) surrounded by CH<sub>4</sub> molecules at a density of 0.58 g/mL. Different cluster sizes were selected to investigate their effects on the reaction mechanism and kinetics. The initial configurations of the Ni<sub>37</sub>, Ni<sub>55</sub>, and Ni<sub>80</sub> clusters were obtained from the Cambridge Cluster Database and selected based on their structures in the lowest energy states, as determined by the Sutton-Chen potential (P. K. Doye & J. Wales, 1998). The simulations were conducted using the ReaxFF 2024.1 software package (Chenoweth et al., 2008; van Duin et al., n.d., 2001), which offers reliable tools for configuring and executing MD simulations with the ReaxFF force field.
The simulations were conducted over 100 ps, with a timestep of 0.25 fs for each run, which allowed the capture of fast reaction events and observation of the intricate dynamics of the systems. A Nosé–Hoover thermostat was employed to ensure temperature control and maintain a constant temperature for the systems throughout the simulations. The systems comprising Ni<sub>55</sub> clusters were simulated at three temperatures: 1,400 K, 1,500 K and 1,600 K. Temperatures of 1400, 1500, and 1600 K were chosen for comparison with previous studies that employed 1500 K (Arifin et al., 2015) to investigate CH<sub>4</sub> reforming.
After the simulations, Ovito (Stukowski, 2009) was used to compute the average surface area of nickel clusters. This is crucial for gaining insight into the reactive surface area required for CH<sub>4</sub> dissociation and subsequent reactions. The activation energies of the reactions were calculated using the Arrhenius equation, as given by Eq. (1).
\[k = Ae^{-\frac{E_a}{RT}} \tag{1}\] where k is the reaction rate constant, A is the pre-exponential factor, \(E_a\) is the activation energy, R is the universal gas constant (8.314 J/mol·K), and T is the temperature (K).
From an analysis of the reaction rates at various temperatures, we plotted the natural logarithm of the reaction rate constant, \(\ln{(k)}\), against the reciprocal of the temperature, 1/T. The slope of this plot, \(-\frac{E_a}{R}\), was then used to determine the activation energy for CH<sub>4</sub> dissociation and H<sub>2</sub> molecule generation.
Results
This section provides results of the simulation, focusing on the mechanisms and efficiencies of \(CH_4\) decomposition and \(H_2\) generation on nickel clusters. First, the impact of the nickel cluster size (\(Ni_{37}\), \(Ni_{55}\), and \(Ni_{80}\)) on the rate of \(CH_4\) decomposition and subsequent generation of \(H_2\) were investigated. Second, the dynamics and reaction mechanisms of the \(Ni_{55}\) cluster were explored, including an examination of the temperature effects and detailed analysis of the simulated atomic interactions.
Effect of Nanocluster Size on CH<sub>4</sub> Decomposition and H<sub>2</sub> Generation
This subsection examines the effects of the nanocluster size on the rate of \(CH_4\) decomposition and \(H_2\) generation. Understanding these size-dependent effects is crucial for maximizing catalytic performance and advancing the development of nanostructured materials for efficient \(CH_4\) conversion and \(H_2\) production.
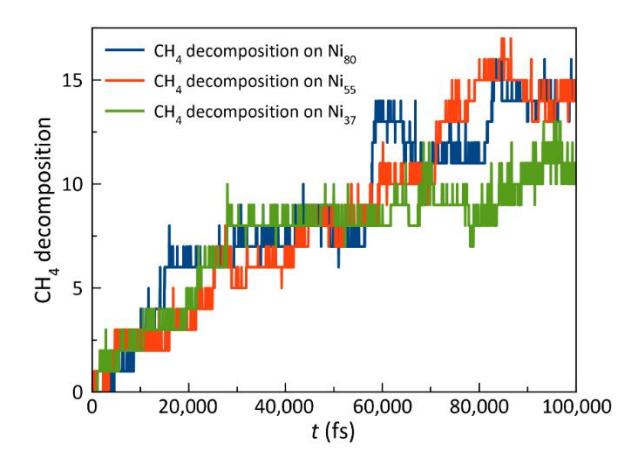
Number of dissociated CH4 molecules on the surfaces of the Ni37, Ni55, and Ni80 clusters over 100,000 fs at T = 1,500 K.
Figure 1 illustrates the dynamic changes in the number of CH4 decomposition events on the surfaces of Ni37, Ni55, and Ni80 clusters at T = 1,500 K. The initial dissociation of CH4 was observed in the Ni37 system, followed by that in the Ni55 and Ni80 systems. Based on the observed trend, the reactivity of nickel nanoclusters decreased as their size increased. Thus, smaller clusters demonstrate a greater initial reactivity. The initial reactivity of the Ni37 clusters was assessed based on the early dissociation of CH4 observed in the simulations. Although this offers a qualitative evaluation, further catalytic activity measurements, including turnover frequency (TOF), will be performed in subsequent studies to enable a quantitative comparison and enhance the understanding of catalytic performance across various cluster sizes. Nevertheless, the Ni37 catalyst exhibited a higher rate of deactivation than those of the Ni55 and Ni80 catalysts. The reaction rate of the Ni37 catalyst significantly decreased after approximately 30,000 fs. In addition, the total number of decomposed CH4 molecules on the Ni37 surface after 100,000 fs was lower than those for the larger Ni55 and Ni80 catalysts.
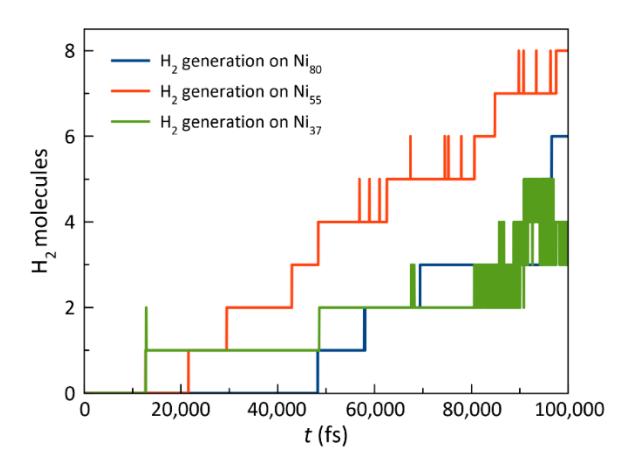
Number of H2 molecules produced on the surfaces of the Ni37, Ni55, and Ni80 clusters over 100,000 fs at T = 1,500 K.
The formation of H2 molecules exhibited a pattern comparable to that of the CH4 decomposition process (see Figure 2), with Ni37 yielding the highest rate of H2 production, followed by Ni55 and Ni80. This suggests that smaller nickel clusters initially facilitate more rapid H2 production because of their higher reactivity. Nevertheless, a notable observation is that the Ni55 system generates a greater number of H2 molecules compared to the Ni37 system within 100,000 fs at a temperature of 1,500 K.
Furthermore, the dynamics of H2 formation over time indicated that the reaction rates for the Ni55 and Ni80 clusters were comparable (as indicated by the slope of the graph, which represents the number of molecules produced per unit time). This indicates that although Ni80 may exhibit a slightly lower initial reactivity, its sustained performance over time is comparable to that of Ni55. Within the time frame of 100,000 fs, the increased total H2 yield of the Ni55 system highlights the potential of this cluster size for efficient H2 production. These findings underscore the need for additional evaluations and longer simulation times to gain a comprehensive understanding of the long-term catalytic performance and stability of nickel clusters of various sizes. Although Ni37 and other smaller clusters may provide rapid initial reaction rates, their rapid deactivation requires a balance between their durability and reactivity. In this regard, Ni55 may represent the optimal size for harmonizing these factors, because it exhibits both high reactivity and sustained catalytic activity, as evidenced by its performance. It is, thus, recommended that future research expand the simulation time beyond 100,000 fs to provide insights into the mechanisms that drive catalyst deactivation and assist in the optimization of nickel clusters for CH4 decomposition and H2 generation over extended periods
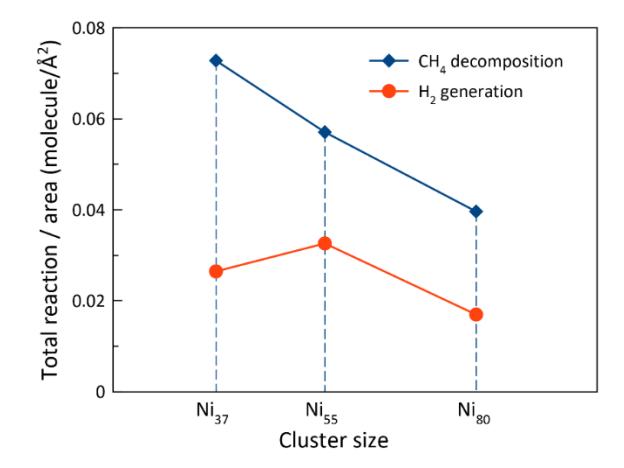
Calculated total reaction/surface area ratio for CH4 decomposition and H2 generation on Ni37, Ni55, and Ni80 clusters over 100,000 fs at T = 1,500 K.
The efficiencies of the catalysts were determined by calculating the total number of CH4 decomposition and H2 formation reactions per unit surface area over 100,000 fs. The system containing Ni37 exhibited the highest number of CH4 decomposition events per unit of surface area during the simulation period, followed by the Ni55 system, with the Ni80 system displaying the lowest number (Figure 3). This suggests that smaller clusters, such as Ni37, have higher surface reactivity for CH4 decomposition at the outset.
Nevertheless, a notable outcome was observed for the formation of the H2 molecules. As illustrated in Figure 3, the system containing the Ni55 nanoclusters yielded the highest number of H2 formation reactions per unit surface area. This implies that although the Ni37 clusters display a higher initial reactivity for CH4 decomposition, the Ni55 clusters exhibit superior efficacy as H2 production catalysts when considering the reaction events per unit surface area.
However, it should be noted that these observations were predicted on a simulation period of 100,000 fs. Additional research is required to assess the long-term stability and efficacy of these catalysts over longer reaction times. Extended simulations can offer a more comprehensive understanding of the durability and sustained catalytic performance of Ni clusters, thereby guaranteeing the reliability of the obtained insights for real-world applications.
CH4 Decomposition and H2 Generation on Ni55 Clusters
Ni55 clusters were used as a model system to elucidate the processes involved in CH4 decomposition and subsequent production of H2. We explored the reaction pathways observed in the simulations, focusing on the complex interactions between the CH4 molecules and the nickel cluster surface. A comprehensive analysis of the chronological order of dissociation events and subsequent formation of H2 molecules was conducted to explain the catalytic processes involved.
Snapshots showing the dissociation of a CH4 molecule on the surface of a Ni55 cluster at T = 1,500 K. The carbon atoms are shown in black, hydrogen atoms in white, and nickel atoms in cyan.
Figure 4 shows the sequence of events that occurred during CH4 dissociation. At t = 3,475 fs, a CH4 molecule in close proximity to a nickel atom (Ni1) can be observed on the surface of the cluster. A hydrogen atom subsequently separates from the CH4 molecule and forms a bond with Ni1 at t = 3,500 fs. At t = 3,650 fs, a hydrogen atom migrated and formed a bond with a nickel atom (Ni2).
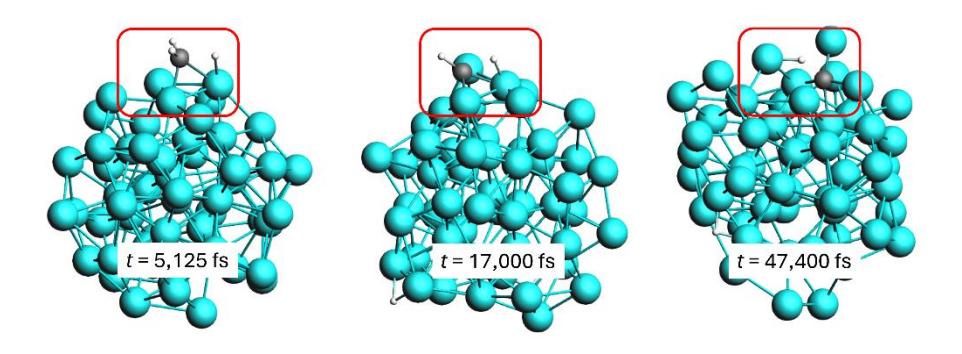
Snapshots showing the dissociation of methyl (left), methylene (center), and methylidyne (right) fragments on the surface of a Ni55 cluster at T = 1,500 K. The carbon atoms are shown in black, hydrogen atoms in white, and nickel atoms in cyan.
Figure 5 shows that the dissociation of a hydrogen atom from the CH3 fragment occurred immediately after the initial dissociation event from the CH4 molecule. The second dissociation of a hydrogen atom occurred at 5,125 fs, and after a considerable period, the third hydrogen atom separated from the CH4 molecule at t = 17,000 fs, leaving behind a CH fragment. At t = 47,400 fs, the CH fragment ultimately underwent decomposition, resulting in the formation of one carbon atom and one hydrogen atom.
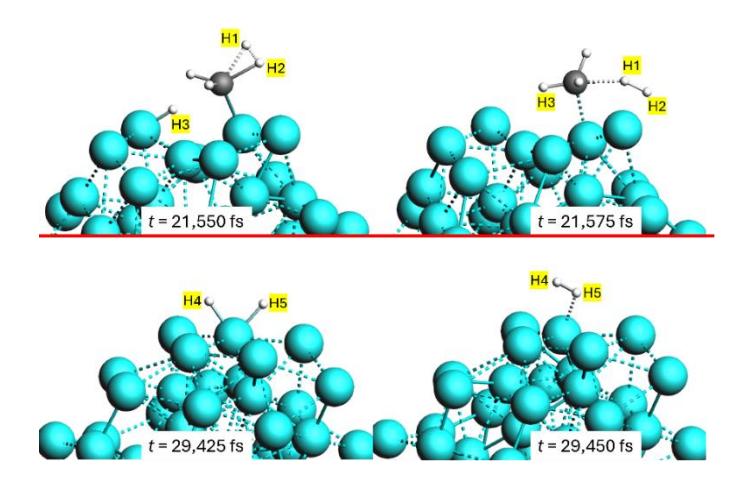
Snapshots showing the mechanism of H2 molecule generation: (top) first H2-production mechanism and (bottom) second H2-production mechanism. The carbon atoms are shown in black, hydrogen atoms in white, and nickel atoms in cyan.
Two mechanisms for H2 molecule formation were observed in the simulations of CH4 reactions on Ni55 clusters, as shown in Figure 6. In the first mechanism (Figure 6, top), a H2 molecule is formed when two hydrogen atoms (H1 and H2) dissociate from a CH4 molecule in the presence of a nickel atom on the cluster surface at t = 21,550 fs. Directly after dissociation, another hydrogen atom (H3) on the nickel surface, close to the site of separation of the initial two hydrogen atoms, combines with the remaining CH2 fragment, generating a CH3 fragment at t = 21,575 fs. This mechanism highlights the immediate interactions and efficient rearrangement of the dissociated hydrogen atoms and CH4 fragments on the cluster surface. In the second mechanism (Figure 6, bottom), two hydrogen atoms (H4 and H5) on the nickel cluster surface combined to form an H2 molecule at t = 29,450 fs. This process occurs without the direct participation of CH4 fragments, suggesting that hydrogen atoms can migrate across the surface and combine to form H2.
The presented mechanisms demonstrate the dynamic nature of the reaction pathways on nickel clusters. The first mechanism involves the simultaneous dissociation and prompt rearrangement of hydrogen atoms and hydrocarbon fragments, resulting in the generation of H2 and CH3 fragments. The second mechanism illustrates the mobility of hydrogen atoms on the nickel surface, enabling them to bond and form an H2 molecule from the original CH4 molecule.
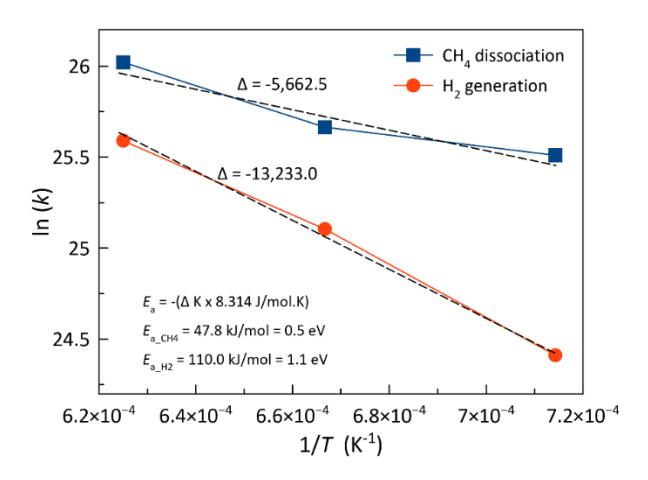
Plots of the natural logarithm of the reaction rate constant, ln (k), versus the reciprocal of the temperature, 1/T, for the dissociation of CH4 molecules and H2 generation on Ni55 clusters. The simulations were performed at T = 1,400 K, 1,500 K, and 1,600 K. The symbols Δ and Ea represent the slopes of the curves and activation energies, respectively.
Our present calculations indicate that the estimated activation energy for CH4 dissociation is 0.5 eV (see Figure 7), notably lower than previously reported values on nickel surfaces (Abild-Pedersen et al., 2005; Arifin et al., 2015; Shen et al., 2016). These findings suggest that Ni clusters exhibit significantly greater reactivity than bulk Ni surfaces. The activation energy for H2 production from CH4 on Ni surfaces can vary depending on the specific conditions and the catalyst employed. As shown in Figure 7, the activation energy for the formation of H2 molecules is 1.1 eV, as determined from our simulations. The substantial energy input required for H2 production via CMD was indicated by its high activation energy. Therefore, it can be inferred that the CMD process requires a significant amount of thermal energy to overcome the energy barrier for H2 formation.
Discussion
Molecular dynamics simulation results for CH4 dissociation on Ni37, Ni55, and Ni80 clusters revealed that smaller nickel clusters exhibited higher reactivity. Consistently, H2 molecules were formed earlier on the Ni37 cluster than on larger clusters. This finding supports those of Niu et al. (Niu et al., 2023), who used density functional theory (DFT) calculations to assess the impact of Ni cluster size on reactivity. However, despite their higher reactivity, smaller clusters such as Ni37 deactivate more rapidly. Therefore, the findings indicate that while smaller clusters may initially exhibit greater
reactivity, they are prone to faster deactivation. This is likely caused by the rapid saturation of active sites or structural modifications that decrease catalytic performance over time. Ni37 clusters are more susceptible to structural rearrangement and sintering because of their smaller size and higher surface energy, which results in a higher rate of deactivation. Furthermore, the increased surface-to-volume ratio of the Ni37 clusters enables the rapid deposition of carbon and surface poisoning, resulting in the loss of active sites. Conversely, the larger Ni55 and Ni80 clusters demonstrate a higher degree of structural stability and a reduced propensity for carbon accumulation, thereby preserving their reactivity for an extended duration.
A similar trend was observed for the production of H2 molecules. Although the Ni37 cluster initially generated H2 at a faster rate, it also underwent a precipitous decline in activity, which was comparable to its mechanism during CH4 dissociation. In contrast, the Ni55 cluster maintained a higher overall yield of H2 molecules despite its delayed initial rate compared to that of Ni37. This indicates that the Ni55 cluster was more effective in maintaining H2 production throughout the simulation period by maintaining a balance between reactivity and stability. The Ni55 clusters attain optimal equilibrium by including sufficient surface reactivity for both CH4 decomposition and sustained H2 production. The balanced surface properties and stability of Ni55 clusters are responsible for their increased efficiency because they can sustain catalytic activity throughout the simulation period. In contrast, the lower initial reactivity of the Ni80 clusters and swift deactivation observed in the Ni37 clusters restrict their overall catalytic efficacy for H2 production within the same time frame. These findings highlight the significant potential of Ni55 nanoclusters as highly efficient catalysts for H2 generation, surpassing the activities of both smaller and larger clusters in terms of reaction occurrence per unit surface area. The exceptional efficiency of the Ni55 clusters may prove indispensable in practical applications that require optimization of H2 yield. Several critical structural and reactive differences can be attributed to the increased overall yield of H2 molecules observed in the Ni55 cluster compared with Ni37 and Ni80. First, the Ni55 clusters exhibited an optimal equilibrium between the structural stability and surface area. Compared to Ni37, the Ni55 cluster has a greater number of active sites, which can facilitate the adsorption and dissociation of CH4. Nevertheless, Ni55 has a more stable and less deformable structure, which enables it to maintain its catalytic activity over time, in contrast to Ni37, which deactivates rapidly because of sintering and carbon deposition. In addition, the Ni55 cluster had a higher density of coordinatively unsaturated sites than Ni80, which is essential for the migration and recombination of hydrogen atoms, thereby increasing the efficacy of H2 formation. Although more stable, Ni80 has a lower yield of H2 per unit surface area because it has fewer highly reactive sites in comparison to its larger size.
Analysis of the atomic trajectories obtained from the MD simulations revealed that the dissociation of CH4 on nickel clusters proceeds in a sequential manner. After the final decomposition step, the carbon atoms were dispersed into the nickel cluster. These findings are consistent with prior research using Ni (110) (Arifin et al., 2025) and Pt7-Ni(110) (Arifin et al., 2024) catalysts, which revealed comparable mechanisms. The sequential nature of the reaction mechanism on nickel clusters is highlighted by the stepwise dissociation of hydrogen atoms and the subsequent decomposition of the CH4 fragment into carbon and hydrogen atoms. The rapid initial dissociation steps were followed by longer periods before further dissociation occurred, suggesting the presence of potential intermediate stabilization and energy barriers that must be overcome for the reaction to progress.
In contrast to the carbon atoms, the dissociated hydrogen atoms exhibited significant mobility on the nickel surface. This mechanism aligns with the findings of previous research, which revealed that hydrogen atoms can migrate across the nickel surface toward specific regions with low energy barriers (approximately 0.3 eV), facilitating their movement on the surface (Iskandarov & Tada, 2017). These findings emphasize the dynamic nature of the interactions of hydrogen with nickel surfaces and highlight the significance of the surface mobility in catalytic processes involving hydrogen. The efficient migration of hydrogen atoms on nickel surfaces can significantly improve the catalytic activity and stability of nickel-based catalysts in a range of applications, such as CH4 reforming and H2 production.
The activation energy of CH4 dissociation on Ni surfaces has been studied extensively. According to Shen et al., the average activation barrier for CH4 dissociation on a Ni(100) surface is approximately 0.90 eV (Shen et al., 2016), aligning with results from DFT calculations. Other researchers have also reported that the initial step of CH4 dissociation on Ni surfaces typically represents the highest barrier, with values around 0.72–0.78 eV on Ni(111) (Arifin et al., 2015) and 0.91 eV on Ni(211) (Abild-Pedersen et al., 2005). These results were higher than the calculated activation energy of CH4 dissociation on the Ni55 cluster (0.5 eV). An important aspect to consider is the change in electronic and other properties as the size of the metal decreases from the bulk material to clusters (Rao et al., 1993). The rise in the core-level binding energy as the metal coverage decreases in clusters indicates a reduction in core-hole screening, which affects their reactivity (Rao et al., 1993). In addition, studies have documented different activation energies for CH4 dissociation on nickel-coated surfaces. Bebelis et al. (Bebelis et al., 2000) reported activation energies ranging between 0.5 and 0.9 kcal/mol for the dissociative adsorption of CH4 on Ni. Notably, even higher activation energies, up to 24 kcal/mol, were estimated by theoretical calculations. Zeng et al. (2021) recently reported that the apparent activation energy for the complete decomposition of CH4 on Ni ranges from 27 kcal/mol to 30 kcal/mol. Meanwhile, Liu et al. revealed a remarkable decrease in the effective barrier for CH4 activation, dropping from 0.9 eV on Ni(111) to a mere 0.15 eV on Ni/CeO2-x(111) (Liu et al., 2016). In addition, the relationship between the catalyst and the support material can affect the activation energy. Li et al. examined the process of CH4 synthesis from syngas on a Ce-doped Ni(111) surface. K. Li et al. identified a specific reaction pathway involving a rate-determining step with an energy barrier of 1.18 eV (K. Li et al., 2016). Further, J. Li et al. (J. Li et al., 2013) examined the impact of a metal-support interface on the dissociation of CH4 and H2 on Ni/γ-Al2O3, emphasizing the significance of this interface in the activation process. Our simulation results also indicated that the formation of H2 molecules requires a higher activation energy than the dissociation of CH4 on the surface of the Ni55 cluster catalyst.
Conclusion
MD simulations were performed using the ReaxFF force field to examine the decomposition of CH4 and generation of H2 on nickel clusters (Ni37, Ni55, and Ni80). The Ni37 clusters exhibited the highest initial reactivity for CH4 dissociation, although they were rapidly deactivated. In contrast, Ni55 and Ni80 maintain consistent reaction rates. The highest number of H2 molecules per unit surface area was produced by Ni55 clusters within 100,000 fs, suggesting their greater efficiency in H2 generation. The dissociation of CH4 on the Ni55 clusters occurred sequentially. Two distinct mechanisms for H2 formation were distinguished: simultaneous dissociation from CH4 and migration and the combination of hydrogen atoms on the cluster surface. The activation energy for CH4 dissociation on Ni55 was 0.5 eV, considerably lower than the reported values for bulk nickel, suggesting that Ni55 has a higher degree of reactivity. The activation energy for H2 formation was estimated to be 1.1 eV. These findings emphasize that Ni55 clusters are among the most effective catalysts for CH4 decomposition and H2 generation because they strike a balance between structural stability and high reactivity. This combination provides new insights into the design of more effective catalytic systems for energy applications, making Ni55 a promising candidate for efficient CH4 conversion and H2 production.
Acknowledgement
This research was supported by a Fundamental Research Grant from the Ministry of Education, Culture, Research, and Technology of Indonesia, under contract number 112/VI.4/PN/2024.
Conflict of Interest Statement
The authors declare that the research was conducted without any financial or commercial relationships that could be construed as a potential conflict of interest.