
1 Introduction
It is well known that diffuse scattering is an important tool for analyzing static and thermal disordered arrangement of atoms in crystals. Anomalously strong and oscillatory diffuse scattering from disordered structure of AgI has been reported by X-ray and neutron scattering experiments [1,2]. The oscillatory diffuse scattering profiles are also observed by neutron scattering experiment even from ordered structure such as AgBr and copper halides [3,4]. The oscillatory profile of diffuse scattering from ordered structure has been explained by including the correlation effects among thermal displacements of atoms up to third nearest neighboring atoms in the crystals. The correlation effects among thermal displacements of atoms are also important in the EXAFS analysis [5,6]. It has been reported that specific features of the vibrational motion of the pairs of atoms in crystals depends on lattice type and chemical bonding [7].
_
In this paper we discuss about structure and diffuse scattering of KBr, with a rocksalt-type structure, by neutron scattering experiments. The diffuse scattering theory with correlation effects among thermal displacements of atoms is applied as background function in the Rietveld analysis of KBr.
2 Experimental
Neutron scattering experiments have been performed on powder KBr at 300 K using HRPD (High Resolution Powder Diffractometer) installed at JRR-3 in Japan Atomic Energy Agency. The sample was put into cylindrical vanadium container of 1.0 cm in diameter. Incident neutron wavelength of 1.823 Å which was monochromatized by Ge (331) was used and the data were collected in the \(2\theta\) range from \(20^{\circ}\) to \(150^{\circ}\) with step angle \(0.05^{\circ}\).
3 Results and Discussion
The diffuse neutron scattering intensity of KBr at 300 K is shown in Figure 1. Several strong Bragg peaks of KBr and diffuse background intensity are observed. The maximum intensity of Bragg line of KBr is about 7000 counts at diffraction angle \(2\theta \sim 45.9^{\circ}\). The typical profile of diffuse scattering intensity which is shown by periodically peaks is observed clearly. The oscillatory diffuse scattering intensity of KBr is constructed by peaks at \(2\theta \sim 30^{\circ}\), \(80^{\circ}\) and \(120^{\circ}\). The crystal structure of KBr is analyzed from the Bragg lines of diffracted intensity using RIETAN-94 [8]. The crystal structure of KBr belongs to NaCl type structure with space group \(Fm\overline{3}m\). The unit cell includes 4 molecules KBr. The crystal contains ordered arrangement and the atomic positions are as follows:
4 K in 4a: 0, 0, 0, 4 Br in 4b: 0.5, 0.5, 0.5.
Table 1 hkl indices and the related d spacing of Bragg lines of KBr.
| \(2\theta\) | hkl | d (Å) | \(2\theta\) | hkl | d (Å) |
|---|---|---|---|---|---|
| 27.70 | 111 | 3.8088 | 91.77 | 333 | 1.2696 |
| 32.08 | 200 | 3.2985 | 102.81 | 440 | 1.1662 |
| 46.01 | 220 | 2.3324 | 109.65 | 531 | 1.1151 |
| 54.55 | 311 | 1.9891 | 111.99 | 442 | 1.0995 |
| 57.19 | 222 | 1.9044 | 121.82 | 620 | 1.0431 |
| 67.10 | 400 | 1.6493 | 129.93 | 533 | 1.0060 |
| 74.06 | 331 | 1.5135 | 132.84 | 622 | 0.9945 |
| 76.33 | 420 | 1.4751 | 146.38 | 444 | 0.9522 |
| 85.20 | 422 | 1.3466 |
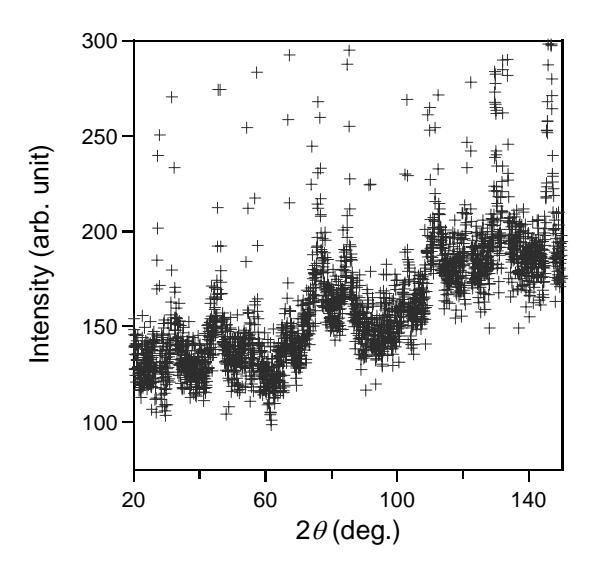
Figure 1 Observed diffuse neutron scattering intensity of KBr at room temperature.
The hkl index of Bragg lines and the related d spacing are presented in Table 1. The structure parameters including lattice constants a and Debye-Waller displacement parameters B for KBr at 300 K which are obtained from refinement analysis of Bragg lines are presented in Table 2.
Table 2 Structural parameter (a: lattice constant and B: Debye-Waller displacement parameter) of KBr at room temperature obtained by Rietveld analysis of powder neutron diffraction data.
| a (Å) | 6.597 | ||
|---|---|---|---|
| \(B_{\rm K}({\rm \AA}^2)\) | 2.070 | ||
| \(B_{\rm Br}({\rm \AA}^2)\) | 1.904 | ||
The mathematical expression to explain the profile of diffuse background scattering intensity is written as follows:
\[I_{\rm B} = kN_{\rm o} \left[ \sum_{i} n_{i} b_{i}^{2} (1 - \exp(-2M_{i})) + \sum_{i} \sum_{j} \sum_{s'} n_{i} b_{i} b_{j} \left[ \exp(-(M_{i} + M_{j})(1 - \mu_{r_{s(i)s'(j)}})) \right] \right]\]
\[-\exp(-(M_i + M_j))]Z_{r_{s(i)s'(j)}}\sin(Qr_{s(i)s'(j)})/(Qr_{s(i)s'(j)})\] \[+\sum_{i}\sigma_i^{\text{inc}} + C\] (1)
where \(N_0\) is number of unit cell in a unit volume. \(n_i\), \(b_i\) and \(\sigma_i^{inc}\) are number of atoms in unit cell, neutron scattering length and incoherent scattering cross section of atom i, respectively. \(Z_r\) is the number of sites belonging to the s'th j-type neighbor around an sth i-type site. Two sites s(i) and s'(j) are apart by distance r. The Debye-Waller factor for atom i, \(\exp(-M_i)\), is equal to \(\exp(-B_i(\sin\theta/\lambda)^2)\). The constant C is added for the corrections of background noise. The correlation effect among atomic thermal displacements \(\mu_r\) is defined as:
\[\mu_{r_{s(i)s'(j)}} = 2 \left\langle \Delta \mathbf{r}_{s(i)} \cdot \Delta \mathbf{r}_{s'(j)} \right\rangle / \left\langle \left\langle \Delta \mathbf{r}_{s(i)}^{2} \right\rangle + \left\langle \Delta \mathbf{r}_{s'(j)}^{2} \right\rangle \right). \tag{2}\] where \(\Delta \mathbf{r}_s\) is the displacement from the mean position caused by thermal vibration. The values of correlation effects among thermal displacements of atoms i and j are 0 in the case of no correlation and \(2(B_i B_j)^{V_2}/(B_i + B_j)\) in the case of perfect correlation [2]. The oscillatory profile of diffuse background intensity is shown by \(\sin(Qr)/(Qr)\).
Table 3 The number of neighboring atoms Z, the values of correlation effects \(\mu\), interatomic distances r and reliability factors R from Rietveld analysis.
| Atomic pairs | r (Å) | Z | μ | |||
|---|---|---|---|---|---|---|
| K-Br | 3.2985 | 6 | 0.657 | |||
| K-K | 4.6648 | 12 | 0.398 | |||
| K-Br | 5.7132 | 8 | 0.150 | |||
| \(R_{\text{wp}} = 8.57\); \(R_I = 3.95\); \(R_F = 2.43\); \(s = 1.36\) | ||||||
Eq. (1) is used as background function in the Rietveld analysis of powder KBr at 300 K. The inter-atomic distances between neighboring atoms in the crystal of KBr are calculated from the refined lattice constants as shown in Table 3. The numbers of neighboring atoms, Z, for first, second and third neighboring atoms in the structure of KBr are 6, 12 and 8, respectively as presented in Table 3.
In the original Rietveld analysis, the Legendre polynomial is used as the background function of diffracted intensity. The profile of the observed
background scattering is approximate by series of mathematical function. The background intensity at step i is calculated as [8]:
\[y_{bi} = \sum_{i=0}^{11} b_j F_j(x_i)\] (3)
where \(F_i(x_i)\) is Legendre polynomial and \(b_i\)'s are coefficients that are optimised in the refinement calculations. The observed background intensity is well fitted by this function. However, there is no physical meaning of the background profile. Equation (1), which replaces the Legendre series as background function in our analysis gives physical meaning of the profile of background scattering intensity. Five parameters in Eq. (1) are refined: the constant \(kN_0\), \(\mu\)'s (the values of correlation effects among first, second and third nearest neighboring atoms) and the constant C. The values of correlation effects \(\mu\) up to third nearest neighboring atoms are obtained from the Rietveld refinement analysis. The obtained values of \(\mu\) together with the reliability factors R are also shown in Table 3. It is seen that the values of correlation effects among thermal displacements of atoms decrease rapidly with increase of inter-atomic distance. The correlation among atomic thermal displacements of far-neighbor atoms usually can be neglected. The profile of the background scattering intensity is mainly determined by the values of correlation effects. In Fig. 2 the calculated intensities using Legendre polynomial and Eq. (1) as background functions are shown together with observed background scattering. The use of Eq. (1) as background function in Rietveld analysis gives similar low reliability R factors to that of Legendre polynomial.
The second term of Eq. (1) is responsible for oscillatory profile of diffuse background intensity. To analyze the influence of correlation effects among atomic thermal displacement to the diffuse scattering profile, we calculated the diffuse background intensity for no correlation case, that is where the values of correlation effects are equal to 0. In this case, atoms vibrate independently which corresponds to Einstein vibration. The second term of Eq. (1) is equal to zero and the diffuse scattering intensity is expressed by first term of Eq. (1) as shown in Fig. 3 by thick solid line. The increase of diffuse background scattering at large scattering angle can be explained by this case, however the oscillatory profile disappears. The oscillatory part of Eq. (1) consists of three parts: first neighbor (K-Br and Br-K), second neighbor (K-K and Br-Br) and third neighbor (K-Br and Br-K) contribution. The calculated diffuse scattering intensities from these contributions are shown in Fig. 3. It is obtained that the diffuse scattering peaks around \(2\theta \sim 80\) and \(120^{\circ}\) are contributed mainly from first and second nearest neighbor atoms. However, the diffuse scattering peak around \(2\theta \sim 30^{\circ}\) might come from the contribution of second and third nearest
neighbor atoms. The observed oscillatory diffuse neutron scatterings of KBr are explained by including the correlation effects among atomic thermal displacements up to third nearest neighbor atoms.
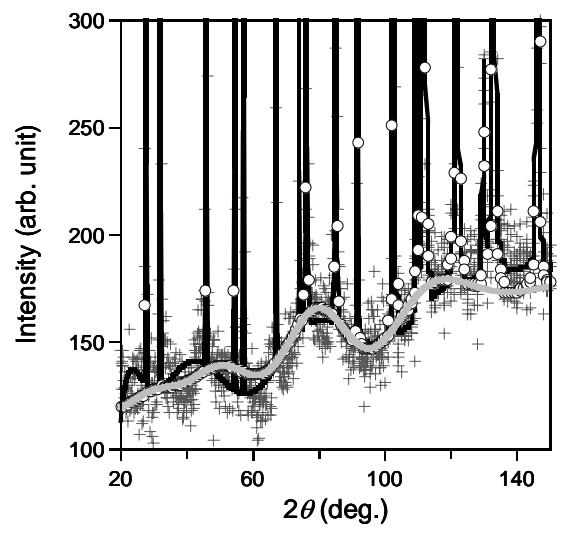
Figure 2 Calculated diffracted intensities of KBr using Legendre polynomial (thick solid line) and Eq. (1) (solid circle) as background function. The observed scattering intensity is shown by plus symbol. The profile of diffuse background scattering is shown by solid gray line.
The values of correlation effects among thermal displacement of atom are very sensitive to inter-atomic distances. Figure 4 shows the values of correlation effects of some cubic crystals obtained by neutron diffraction measurements. The horizontal axis is inter-atomic distance relative to the lattice constants of the crystals. The values of correlation effects for first nearest, second nearest and third nearest neighboring atoms are 0.6 − 0.8, 0.3 − 0.5 and 0.1 − 0.2, respectively. The correlation among thermal displacements of atoms for r/a > 1.0 relatively are very small and might be neglected. The contribution of third nearest neighboring atom to the diffuse scattering profile usually appears at relatively low scattering angle since the oscillatory form is expressed by sin(Qr)/(Qr) in Eq. (1).
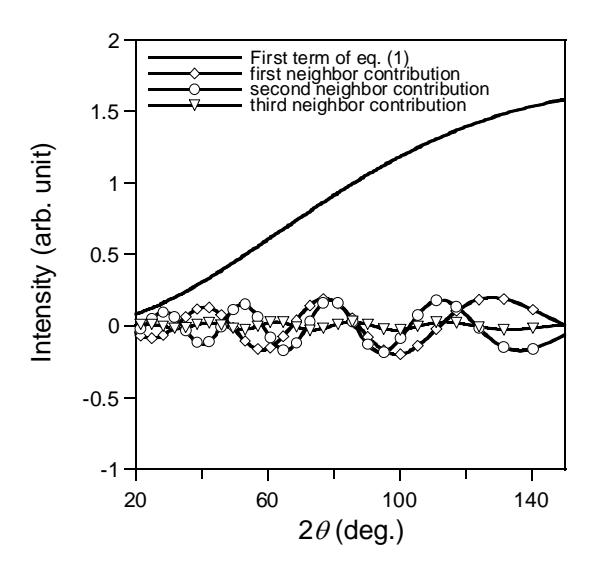
Figure 3 Calculated contribution of diffuse scattering intensity of KBr.
Yoshiasa et al. [7] studied the MSD (mean square displacements) and DCF (displacements correlation function) of cubic A<sup>N</sup>B<sup>8-N</sup> crystals by diffraction measurements. The values of DCF and MSD are important factors in the analysis of lattice dynamics of crystals by EXAFS (Extended X-ray Absorption Fine Structure) method [10]. The values are also can be used to predict effective potential and local force constant between atoms in crystals [11]. The ratio of DCF and MSD is similar to the correlation effects in our analysis. According to Yoshiasa et al. [7], the value of the DCF/MSD ratio is related to the type of atomic bonding and the coordination number. The correlation effects for crystals with cation/ anion in octahedral coordination are relatively smaller than those in tetrahedral coordination. The values of DCF/MSD for octahedrally ionic bonding crystals are usually less than that for tetrahedrally covalent bonding crystals.
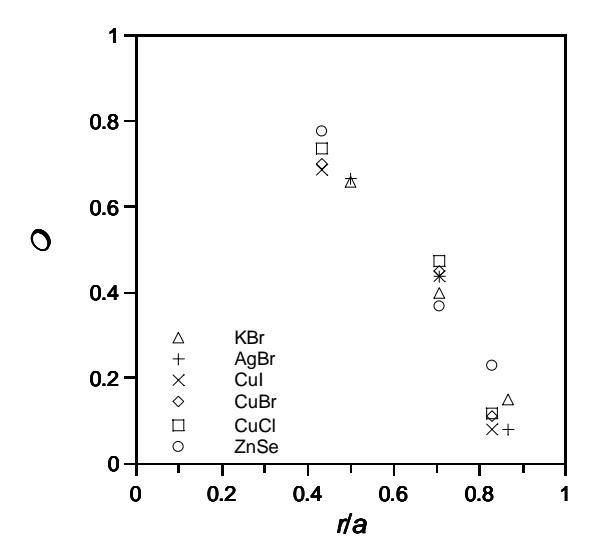
Figure 4 Correlation effects among thermal displacements of atoms in some cubic crystals (AgBr [3], copper halides [4], ZnSe [9] and KBr [this work]). The horizontal axis is the inter-atomic distances relative to the lattice constants.
4 Conclusion
The diffuse neutron scattering profile of KBr at room temperature is explained by including the correlation among thermal displacements of atoms up to third neighbor atom in the crystal KBr. The values of correlation effects decrease with inter-atomic distance. The diffuse scattering theory including correlation effects is applied as background function in Rietveld analysis of diffracted intensity of powder KBr as well as other cubic crystal with ordered arrangement of atoms [4,9].
5 References
- [1] Hoshino. S., Crystal Structure and Phase Transition of Some Metallic Halides (IV) On The Anomalous Structure of α-AgI, J. Phys. Soc. Jpn., 12, 315 – 326, 1957.
- [2] Sakuma, T., Diffuse Scattering of α-AgI, J. Phys. Soc. Jpn., 61, 4041 4048, 1992.
- [3] Arai, M., Shimoyama, T., Sakuma, T., Takahashi, H., Ishii, Y., Correlation effects between thermal displacements of atoms in diffuse scattering from AgBr, Solid State Ionics, 176, 2477 – 2480, 2005.
- [4] Sakuma, T., Shimoyama, T., Basar, K., Xianglian, Takahashi, H., Arai, M., Ishii, Y., Correlation effects between thermal displacements of atoms in copper halides, Solid State Ionics, 176, 2689 – 2693, 2005.
- [5] Beni, G., Platzman, P. M., Temperature and polarization dependence of extended x-ray absorption fine structure spectra, Phys. Rev., B14, 1514 – 1518, 1976.
- [6] Böhmer, W., Rabe, P., Temperature dependence of the mean square relative displacements of nearest-neighbour atoms derived from EXAFS spectra, J. Phys. C: Solid State Phys., 12, 2465 – 2474, 1979.
- [7] Yoshiasa, A., Koto, K., Maeda, H., Ishii, T., The Mean-Square Relative Displacement and Displacement Correlation Functions in Tetrahedrally and Octahedrally Coordinated AN B8−N Crystals , Jpn. J. App. Phys., 36, 781 – 784, 1997.
- [8] Izumi, F., Rietveld analysis programs RIETAN and PREMOS and special application, The Rietveld Method, R. A. Young (ed.), Oxford University Press, 236 – 253, 1993.
- [9] Basar, K., Siagian, S., Xianglian, Sakuma, T., Takahashi, H., Igawa, N., Correlation effects among atomic thermal displacements in oscillatory diffuse neutron scattering of ZnSe, Nucl. Inst. Met. Phys. Res. A600 (2009) 237.
- [10] Fornasini, P., Study of lattice dynamics via extended x-ray absorption fine structure, J. Phys.: Condens. Matter, 13, 7859 – 7872, 2001.
- [11] Hung, N. V., Trung, N. B., Hung, L. H., Anharmonic effective potential, local force constant and correlation effects in XAFS of BCC crystals, Adv. in Nat. Sci., 7, 13 – 30, 2006.