
1 Introduction
Recently, the use of synthetic polymers as plastics has been increasing very rapidly, but the plastic waste of synthetic polymers is a serious problem for the environment because most synthetic polymers cannot be decomposed by microorganisms. New techniques and processes are focused on obtaining biodegradable polymers since they are environmentally friendly. One of the alternative techniques is synthesis of new biodegradable synthetic polymers by using natural resources as raw materials. Biodegradable polymers generally contain hydrolysable bonding in the backbone chains. The polymer bonding that is easily hydrolyzed by acid or enzymes produced by microorganisms consists of polar functional groups such as ester, amide, and peptide groups [1,2].
Polyurethane is a synthetic polymer used in various applications because it has the advantage of synthesizing with different polymer materials that have diverse mechanical and physical properties due to various polyol and di-isocyanate compounds that can be used in synthesis. It is possible to prepare polyurethane in different shapes, such as film or hydrogel, with different structures and properties, such as water permeability, biocompatibility, and biodegradability, according to types of reactants, component ratio, and reaction conditions [3]. Generally, polyurethane is a macromolecule containing functional groups of urethane (-NHCOO-) in the backbone chain. This functional group is formed by condensation polymerization of compounds containing isocyanate and hydroxyl groups or between di-isocyanate compounds with polymers containing hydroxyl groups on the end side chain, which are called polyol compounds [4-6]. Some types of polyol compounds used as a prepolymer to prepare polyurethane are polyethers, such as poly(tetra-methylene glycol), poly(propylene glycol), poly(ethylene glycol), and polyesters containing terminal hydroxyl groups such as poly(ethylene adipate) and poly(propylene adipate), and glycerol adipate [7,8]. Di-isocyanate compounds mostly used to prepare polyurethane are hexamethylene-1,6-di-isocyanate (HMDI), 4,4-methylen-bis phenyl isocyanate (MDI), and toluenil-2,4-di-isocyanate (TDI). Polyurethane prepared by polymerization of aromatic di-isocyanate, such as MDI and TDI, has a hard and stiff molecule structure and high thermal and mechanical properties [5,6].
Polyurethane is now widely used in various applications, for example, as raw material for foam, paints, coatings, elastomers, as a packaging material, as well as for biomedical and automotive uses. One of the raw materials in the manufacture of polyurethanes is vegetable oil. Among the various types of vegetable oils, palm oil has great potential to be further developed for raw materials of biodegradable polymers because Indonesia is currently the world's largest producer of CPO (crude palm oil) in the world and has the largest land area for palm oil cultivation in the world [9,10]. In addition, palm oil is a source of renewable raw materials, which has more value than exhaustible raw materials like those derived from petroleum (fossil). Moreover, from the side of the resulting product, modification of palm oil produces a polymer with various applications that can give additional value to agricultural production.
The long-term goal of this research is to get a plastic material that is easily decomposed by microorganisms in nature, either by chemical synthesis or by modification of existing polymers, as well as to examine the relationship between the chemical structure and the polymer properties, including their biodegradability. For this purpose, the present research was focused on the preparation of biodegradable polyurethanes as a plastic material for wide use in daily life by using palm oil as a source of polyols. This study was carried out in two stages. The first stage focused on the synthesis of a prepolymer or polyol
through modification and purification of palm oil on various parameters. The second stage consisted of the synthesis of poly(urethane) through polymerizing polyol and di-isocyanate with or without addition of a chain extender.
In this study, 9-ethoxy-1,10-octadecanediol as the diol compound obtained by modification and purification of oleic acid of palm oil, poly(urethane) and poly(urethane-urea) obtained by polymerizing polyol and di-isocyanate with ethylene diamine used as chain extender, were characterized. All compounds were characterized by chemical structure analysis (FTIR and 1 H NMR), supported by analysis of iodine number, acid number, and hydroxyl number determined by titration, and analysis of thermal properties and intrinsic viscosity especially for poly(urethane) and poly(urethane-urea).
2 Experimental
2.1 Materials
Oleic acid, acetic acid, formic acid, and hydrogen peroxide, aluminum trichloride (AlCl3) were obtained commercially from E-Merck Chem.Co. Sodium boron tetra-hydride (NaBH4) and 4,4-methylen-bis phenyl isocyanate (MDI) were obtained commercially from Aldrich Chem. Co. All above mentioned chemicals were used as received without further purification.
2.2 Modification of Oleic Acid
2.2.1 Epoxydation of Oleic Acid
A mixture of oleic acid (0.175 mole) and acetic acid (0.2 mole) in 2.5 g toluene as solvent was heated at a temperature of 55 °C. Another solution was prepared by mixture of 0.2 mole H2O2 and 0.05 mL concentrated sulfuric acid. This solution was added drop-wise to a solution containing oleic acid and then stirred for 12 hours at a temperature of 55 °C to produce phase separation between the organic and aqueous phase, after which the aqueous phase was separated by sedimentation. To neutralize the residual of sulfuric acid, the mixture was washed with distilled water, until it had a neutral pH, and decanted to remove water. The compound produced by epoxidation of oleic acid was dried in a vacuum oven to remove the residual water [11,12].
2.2.2 Ring-opening of Epoxide Oleic Acid
A mixture of ethanol (3.0 mole), p-toluene sulfonate (5% w/w of epoxide oleic) as catalyst in 5.0 g toluene was stirred for 15 minutes at 60 °C, and then the epoxide oleic (1.0 mole) was added to this solution and stirred for three hours.
The mixture was neutralized with distilled water and the residual water was removed by decantation and dried in a vacuum oven to produce an ester of oleic acid [11,13].
2.2.3 Reduction of Ester Oleic Acid
In a separate glass, 0.225 mole NaBH4 and 0.084 mole AlCl3 were dissolved in diethyl glycol-dimethyl ether (diglyme) and stirred with a magnetic stirrer. The solution of NaBH4 was heated at a temperature of 50 °C, and then 0.4 mole ester that had been prepared previously was added into the NaBH4 solution. The AlCl3 solution was then added drop-wise to the solution and stirred continually for three hours. After the reaction was complete, the mixture was cooled to room temperature, and the diol compound formed was separated and dried in a vacuum oven [14].
2.3 Preparation of Poly(urethane-urea)
The preparation of poly(urethane) was carried out with various ratios of –NCO functional groups of MDI to -OH functional groups of diol compound without catalyst [15]. In a separate glass, monomer of 4,4'-methylene-bis (phenyl isocyanate) (MDI) and the prepared diol compound was dissolved in N,Ndimethyl formamide (DMF) and heated at 85 °C. The diol compound solution was poured into a glass three-neck, after which the solution of MDI was added under continually stirring for 15 minutes. In the last stage, ethylene diamine was added to the reaction mixture as chain extender and stirred for one hour at 85 °C. After reaction was complete, the mixture was cooled to room temperature and precipitated in a mixture of water/ethanol (80/20 v/v). The precipitate obtained was washed with ethanol and dried in a vacuum oven at 60 °C for 2-3 days and then compression-molded with a hot press at 150 °C for 5 minutes to form a thin film.
2.4 Characterization of Polymers
FTIR (Shimadzu 5800) was used to characterize the chemical structure of the polymers. The sample used in FTIR was in KBr pellet form. The 1 H NMR measurement was recorded at 500 MHz on a JOEL JNM-FX 400 spectrometer. The sample concentration used in this measurement was typically 0.1% wt/vol. The NMR spectrum of the polymers was recorded at 25-30 °C with CDCl3 as solvent. Tetramethylsilane (TMS) was used as a reference. A differential thermal calorimeter (DTA/TGA) (General Du Pont 2000) was used to determine the thermal behavior of the polymers. All scans were carried out from room temperature to 550 °C at a heating rate of 10°C min-1. The molecular weights of the polymers were determined by viscometer.
The oxirane oxygen number of the compounds was determined by titration according to AOCS method Cd 9-57. A sample of 0.3-0.5 g was added to a solution of 10 mL glacial acetic acid or chlorobenzene (for epoxy resin) and 0.5 ml violet indicator was added. The solution was then titrated with 0.1 M hydrogen bromide (HBr) to form a blue-green solution. The oxirane oxygen number can be determined by Eq. (1):
Oxirane oxygen number (%) = \[\frac{BxMx1.60}{m}\] (in g sample) (1)
where B = HBr volume required in sample solution titration (mL), M = HBr concentration used in titration (M), and m = mass of sample used in titration (g).
The acid number of the samples was determined by titration according to the standard method (ISO 660). A sample of 10-20 g was added to a solution of 50 mL neutral alcohol 95% (neutralized with NaOH and checked by phenolphthalein indicator). This solution was heated for 10 minutes, after which 3 drops of phenolphthalein indicator were added. The solution was then titrated with 0.1 M potassium hydroxide (KOH) to form a pink solution. The hydroxyl number can be determined by Eq. (2):
Acid number = \[\frac{AxMx56.1}{m}\] (2)
where A = KOH volume required in sample solution titration (mL), M = KOH concentration used in titration (M), and m = mass of sample used in titration (g).
The hydroxyl number of the copolymers was determined by titration according to AOAC Official Method 965.32. A sample of 0.5 g was added to a solution of 4 mL acetic anhydride in pyridine, which was prepared in a mixture of 127 mL acetic anhydride and 1000 mL pyridine. This solution was heated at 98 °C for 2 hours, after which 6 mL distillated water and 3 drops of phenolphthalein indicator were added. The solution was then titrated with 0.1 M potassium hydroxide (KOH). The hydroxyl number can be determined by Eq. (3):
\[Hydroxy number = \frac{(B-A)xMx56.1}{m}\] (3)
where B = KOH volume required in standard solution titration (mL), A = KOH volume required in sample solution titration (mL), M = KOH concentration used in titration (M), and M = MSOH concentration used in titration (g).
The iodine number is the amount of iodine bound by 100 g of fat and indicates the number of double bonds or unsaturated bonds. The iodine value of the sample was determined by titration according to AOAC Official Method 920.158. A sample of 0.1-0.5 g was dissolved in chloroform or tetrachloromethane, 25 mL iodine was added, after which it was placed in a dark room. 10 mL solution of KI 15%, 100 mL distillated water, and starch indicator were added to this solution, after which it was titrated with a solution of Na<sub>2</sub>S<sub>2</sub>O<sub>3</sub> 0.1N until the color of the solution changed from blue to clear. The same method was performed for a blank solution. The iodine value can be determined by Eq. (4):
Iodine value = \[\frac{(B-A)xNx12.69}{m}\] (4)
where \(B = Na_2S_2O_3\) volume required in blank solution titration (mL), \(A = Na_2S_2O_3\) volume required in sample solution titration (mL), \(N = Na_2S_2O_3\) used in titration (N), and \(M = Ma_2S_2O_3\) used in titration (g).
3 Results and Discussion
Polyol of 9-ethoxy-1,10-octadecanediol obtained by modification of palm oil was prepared in three stages namely: epoxidation of oleic acid using peroxyacetic acid and sulfuric acid as catalyst to produce an epoxide of oleic acid; ring-opening of epoxide on oleic acid using ethanol to yield a 9 or 10 ethoxy ester of oleic acid; and subsequently reduction of the ethoxy oleic acid ester with sodium boron-hydride as reductor and hydrogen chloride as catalyst to yield a diol compound of the bio-substrate.
\[\text{[rumus tidak dapat ditampilkan dengan baik — lihat PDF asli]}\]
Figure 1 Epoxidation reaction of oleic acid.
Epoxidation of the oleic acid or palm oil was done by using peroxyacetic acid and sulfuric acid as catalyst at 55 °C for 12 hours to form an epoxide of oleic acid (3-octyl oxirane octanoic acid) with a maximum yield of 93% (0.175 mole in 2.5 g toluene as solvent, 0.2 mole acetic acid, 0.2 mole \(H_2O_2\) 35%, 0.05 mL sulfuric acid) (Figure 1).
The resulting epoxide of oleic acid was analyzed through the change in iodine number, oxygen-oxirane number, and retention time (GC), functional groups (FTIR), and chemical shift (<sup>1</sup>H NMR).
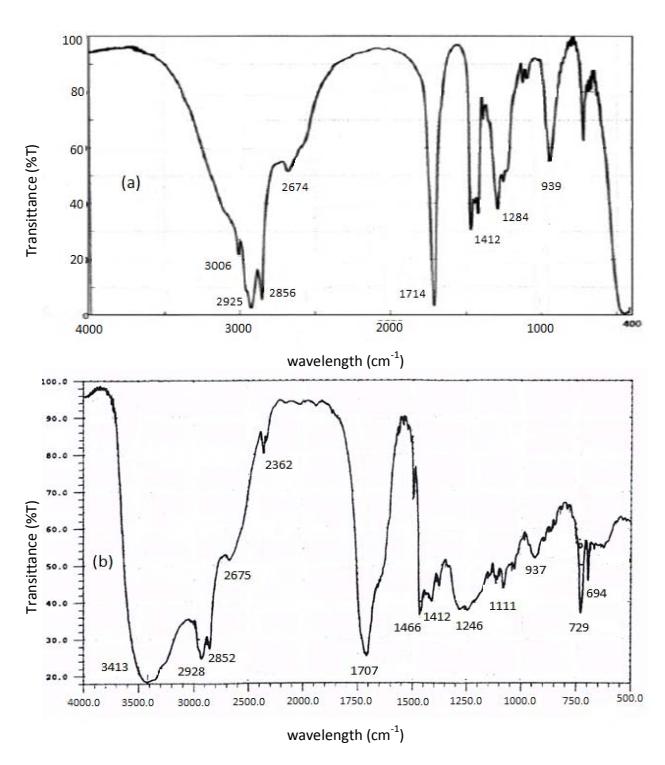
Figure 2 FTIR spectra of (a) oleic acid and (b) oleic acid epoxide.
To prove the presence of oxirane ring of oleic acid epoxide, a comparison between the FTIR spectra of the oleic acid and the epoxide oleic acid is shown in Figure 2 while the \(^1\)H NMR spectra are shown in Figure 3. Chemical shifts \((\delta, ppm)\) \(^1\)H-NMR of oleic acid (500 MHz, CDCl<sub>3</sub>): \(\delta\) 0.9(3H, -CH<sub>3</sub>), 1.3(20H, -CH<sub>2</sub>-), 1.6(2H, -CH<sub>2</sub>COO<sub>2</sub>H), 2.0(4H, -CH<sub>2</sub>CH=CHCH<sub>2</sub>-), 2.3(2H, -CH<sub>2</sub>COO<sub>2</sub>H), 5.4(2H, -CH=CH-), and 7.3(1H, -OH). \(^1\)H-NMR of epoxide oleic acid (500 MHz, CDCl<sub>3</sub>): \(^1\)H-NMR (500 MHz, CDCl<sub>3</sub>): \(^-\delta\) (ppm) 0.9(3H, -CH<sub>3</sub>), 1.2-1.5(20H, -CH<sub>2</sub>- and 4H, -CH<sub>2</sub>CHOCCH<sub>2</sub>-), 1.7(2H, -CH<sub>2</sub>COO<sub>2</sub>H), 2.3(2H, -CH<sub>2</sub>COOH), 3.6(2H, -CHOCH-), and 7.4(1H, -OH).
The FTIR peaks of the epoxide oleic acid indicate the appearance of an absorption peak at 694 cm<sup>-1</sup>, which belongs to the oxirane ring. The intensity of the absorption peaks at wavenumber 939 cm<sup>-1</sup> for =CH<sub>2</sub> groups of oleic acid decreased significantly after epoxidation, but the intensity of the absorption peaks at wave number around 3250-3600 cm<sup>-1</sup> indicating epoxy groups increased and widened, and also the presence of chemical shifts of -CH=CH-protons and -CH<sub>2</sub>-CH=CH-CH<sub>2</sub>- groups in the oleic acid at 5.33-5.34 ppm and 2.00 ppm (Figure 3a) became 3.47-3.57 ppm and 1.47 ppm after epoxidation, i.e.-CH-O-CH- protons and -CH<sub>2</sub>-CHOCH-CH<sub>2</sub>- groups, respectively (Figure
3b) [16]. In addition, the iodine number, oxygen-oxirane number, and retention time for oleic acid were 84.9 g I<sub>2</sub>/100 g, 0.0 g O/100g, and 5.5 minutes, respectively. After epoxidation, the resulting compound had iodine number, oxygen-oxirane number, and retention time for oleic acid at 2.17 g I<sub>2</sub>/100 g, 5.42 g O/100g, 7.3 minutes, respectively. The iodine number of the prepared epoxide was not equal to zero, because the epoxidation of oleic acid is not completed at optimum conditions (12 hours) and subsequently a small fraction of the double bonds still exists in the oleic acid molecule. These results indicate the change of oleic acid into 3-octyl oxirane octanoic acid after epoxidation.
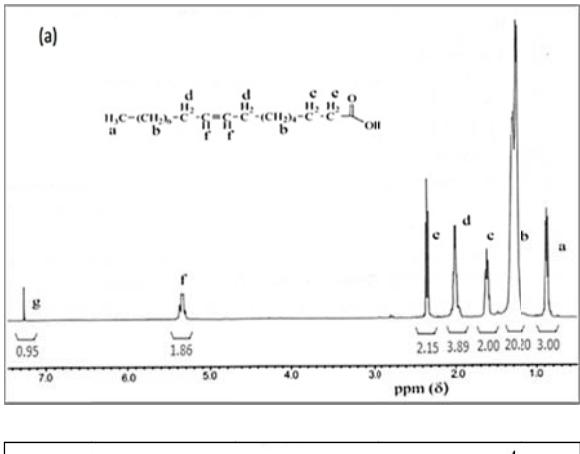
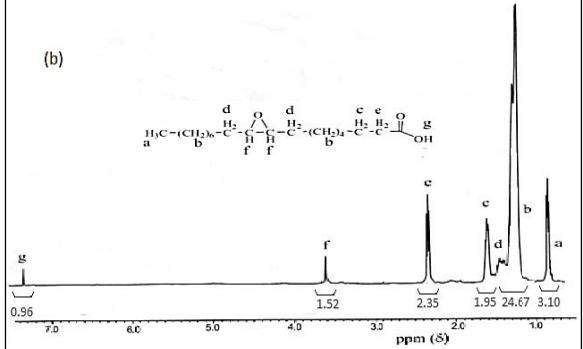
Figure 3 <sup>1</sup>H NMR spectra of (a) oleic acid and (b) oleic acid epoxide.
The ring-opening of 3-octyl oxirane octanoic acid was done by using ethanol and p-toluene sulfonate as catalyst at 60°C for 3 hours to produce an ester of oleic acid, i.e. 9-ethoxy-10-hydroxy-octadecanoic ethyl ester (1.0 mole 3-octyl oxirane octanoic acid, 3.0 mole ethanol in 5 g toluene, 5% (w/w) p-toluene sulfonate of mass oleic acid epoxide used) (Figure 4).
\[\text{[rumus tidak dapat ditampilkan dengan baik — lihat PDF asli]}\]
Figure 4 Ring-opening reaction of oleic acid epoxide.
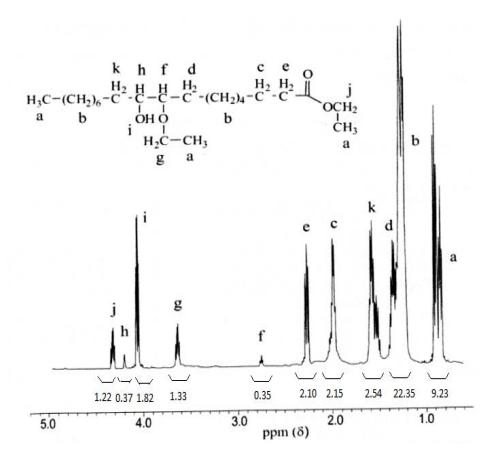
Figure 5 <sup>1</sup>H NMR spectrum of 9-ethoxy-10-hydroxy-octadecanoic ethyl ester (oleic acid ester).
This interpretation was supported by the chemical shift analysis by <sup>1</sup>H NMR (Figure 5). Chemical shifts (δ, ppm) <sup>1</sup>H-NMR of 9(10)-ethoxy-10(9)-hydroxy-octadecanoic ethyl ester (500 MHz, CDCl<sub>3</sub>): ~δ (ppm) 0.9(9H, -CH<sub>3</sub>), 1.3(20H, -CH<sub>2</sub>-), 1.4(2H, -CH<sub>2</sub>CHOCH<sub>2</sub>CH<sub>3</sub>), 1.5(2H, -CH<sub>2</sub>CHOH), 1.6(2H, -CH<sub>2</sub>CH<sub>2</sub>CO<sub>2</sub>CH<sub>2</sub>CH<sub>3</sub>), 2.3(2H, -CH<sub>2</sub>CO<sub>2</sub>CH<sub>2</sub>CH<sub>3</sub>), 2.8(1H, -CHO CH<sub>2</sub>CH<sub>3</sub>), 3.7(2H, -OCH<sub>2</sub>CH<sub>3</sub>), 4.1(H, -OH), 4.3(H, -CHOH), and 4.5(2H, -CO<sub>2</sub>CH<sub>2</sub>CH<sub>3</sub>) [17]. The appearance of new peaks in the 9-ethoxy-10-hydroxy-octadecanoic ethyl ester at a chemical shift of 0.80-1.00 ppm (multiplet) can be seen as three triplet peaks of -CH<sub>3</sub>, derived from two methyl protons in the ethoxy group and protons in the methyl group (-OCH<sub>2</sub>CH<sub>3</sub> and -CH<sub>3</sub>), 4.35 ppm from ethyl protons in the ester group (-COOCH<sub>2</sub>CH<sub>3</sub>), and 3.62-3.64 ppm from ethyl protons in the ether group (-COCH<sub>2</sub>CH<sub>3</sub>).
Reduction of oleic acid ester was done by using sodium boron-hydride (NaBH<sub>4</sub>), aluminum chloride (AlCl<sub>3</sub>), and hydrogen chloride as catalyst at 50 °C for 3 hours because hydrogen chloride is easier to handle and is not reactive to water (0.225 mole NaBH<sub>4</sub> and 0.084 mole AlCl<sub>3</sub> in ethylene glycol-dimethyl ether as solvent, in a separate glass, respectively, and 0.4 mole oleic acid ester) (Figure 6).
\[HO\] \(H\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H_5\) \(OC_2H\)
Figure 6 Reduction reaction of oleic acid ester.
The FTIR spectra of the products have some common peaks at 2927 and 2854.5 cm<sup>-1</sup> (methylene asymmetric stretching) and 1465.8 cm<sup>-1</sup> (-CH<sub>2</sub>- bending vibration). To prove the presence of hydroxyl groups of 9-ethoxy-1,10-octadecanediol, a comparison between 9-ethoxy-10-hydroxy-octadecanoic ethyl ester and 9-ethoxy-1,10-octadecanediol FTIR spectra is shown in Figure 7.
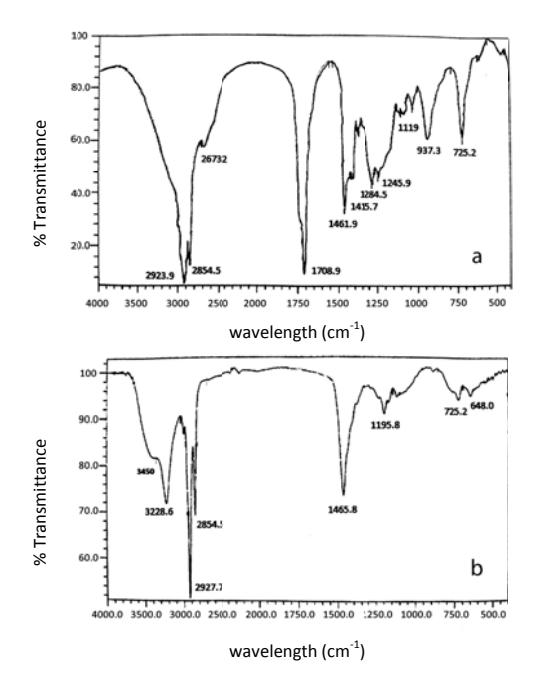
Figure 7 FTIR spectra (a) 9-ethoxy-10-hydroxy-octadecanoic ethyl ester and (b) 9-ethoxy-1,10-octadecanediol.
The FTIR peaks of the oleic acid epoxide indicate the disappearance of the absorption peak at 694 cm<sup>-1</sup> that belongs to the oxirane ring. The intensity of some absorption peaks, for example –OH groups (3413 cm<sup>-1</sup>), decreased or disappeared (Figure 7a). In addition, the increase of absorption peak intensity at 1708 cm<sup>-1</sup>, derived from carbonyl and secondary alcohol groups, indicate the occurrence of ring-opening of the oleic acid epoxide to form oleic acid ester (9(10)-ethoxy-10(9)-hydroxy-octadecanoic ethyl ester).
The FTIR spectra of 9-ethoxy-1,10-octadecanediol exhibit peak intensity of hydroxy groups at 3450 cm<sup>-1</sup> (Figure 7b) and a new absorption peak appeared around 3228.6 cm<sup>-1</sup>, which is a typical absorption peak of asymmetric stretching of the -OH group (Figure 7b), whereas new absorption peaks disappeared around 2673 cm<sup>-1</sup> and 1708.9 cm<sup>-1</sup>, which are typical absorption peaks of the – COOH or the –COOCH- group and stretching vibration of the –C=O group, respectively. These results indicate the change of 9-ethoxy-10-hydroxy-octadecanoic ethyl ester to produce 9-ethoxy-1,10-octadecanediol. This result was supported by analysis of the hydroxyl numbers before and after reduction of oleic acid ester determined by titration. The hydroxyl number for 9-ethoxy-10-hydroxy-octadecanoic ethyl ester is 145.6 mg KOH/g, and after reduction by using NaBH<sub>4</sub>/AlCl<sub>3</sub>, and HCl produces 9-ethoxy-1,10-octadecanediol, which has a hydroxyl number of 322.0 mg KOH/g.
3.1 Poly(urethane) and Poly(urethane-urea)
Poly(urethane) as a prepolymer was obtained through polymerization of 9-ethoxy-1,10-octadecanediol and 4,4-methylen-bis phenyl isocyanate (MDI) under nitrogen atmosphere with a mole ratio of –NCO functional group of MDI to -OH functional group of the diol compound 1.3/1(mole/mole) in the absence of a catalyst at 85°C for 15 minutes, whereas poly(urethane-urea) was prepared by addition of ethylene diamine (EDA) as chain extender to poly(urethane) with a mole ratio of –NCO/-OH/EDA = 1.3/1.0/1.0. The mechanism of polymerizing polyol and 4,4-methylen-bis phenyl isocyanate with ethylene diamine used as chain extender to produce poly(urethane-urea) is shown in Figure 8.
\[\begin{array}{c} \text{OC}_{2}\text{H}_{5} \\ \text{HO} \\ \text{CH} \\ \text{CH}_{2}\text{C} \\ \text{O} \\ \text{CH}_{2}\text{C} \\ \text{O} \\ \text{CH}_{2}\text{C} \\ \text{O} \\ \text{CH}_{2}\text{C} \\ \text{O}_{7}\text{CH}_{3} \\ \text{Diol oleic} \\ \end{array}\]
Figure 8 Mechanism of polymerizing 9-ethoxy-1,10-octadecanediol and 4,4-methylen-bis phenyl isocyanate with ethylene diamine used as chain extender.
Poly(urethane) and poly(urethane-urea) were characterized by analysis of functional groups (FTIR), chemical shift (<sup>1</sup>H NMR), molecular weight (viscometer), and thermal properties (TGA). The sharp absorption peaks around 3309.6 cm<sup>-1</sup> and 1739.7 cm<sup>-1</sup> are typical absorption peaks for functional groups of –NH and –C=O derived from the urethane group, respectively.
The other absorption peaks indicate the formation of a urethane bond between the –OH functional group of the diol compound with the –NCO functional group of MDI. The absorption peaks at 1018.3 cm<sup>-1</sup> are derived from stretch vibration of C-O-C, those at 1307.6 cm<sup>-1</sup> and 1238.2 cm<sup>-1</sup> from stretch vibration of –CN and bending vibrations of –NH of amide groups (Figure 9a). Both poly(urethane) and poly(urethane-urea) have similar functional groups because the new chemical structure still has the same or a repetition of chemical structures that already existed in the monomers so that the FTIR spectra do not appear specific around newly formed absorption peaks.
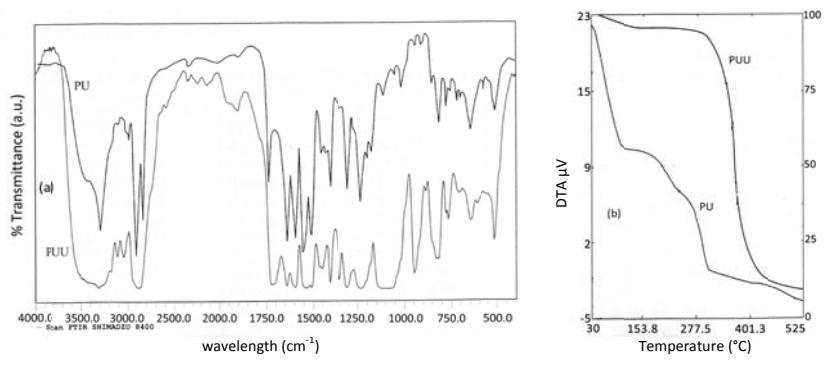
Figure 9 (a) FTIR spectra of PU and PUU and (b) TGA thermogram of PU and PUU.
Thermal analysis using TGA shows the presence of the fourth stage of thermal decomposition in poly(urethane) (Figure. 9b). The first stage transition about 30-100 °C is the release of side chains of the polymer such as ethanol and octane groups with the remaining material at 60%. The next two stages between 170-255.5 °C and 260-325 °C, are the decomposition of urethane bonds in the polymer by releasing carbon dioxide and ammonia with the remaining material at 12%. The final stage is the transition between 325 and 525 °C with the remaining carbon at 6.5%. However, poly(urethane-urea) shows two stages of thermal decomposition. The first-stage transition at about 30-345 °C is the release of side chains of the polymer such as water or ethanol with the remaining material at 81.6%. The second stage occurs between 350 and 401 °C, and is the decomposition of urethane bonds in the polymer by releasing carbon dioxide and ammonia and the decomposition of side chains such as octane groups with the remaining material at 17.5%. The final stage transition occurs
between 401-525 °C with the remaining carbon at 7.9%. The above results show that the thermal stability of poly(urethane-urea) is higher than that of poly(urethane) because poly(urethane-urea) has a longer chain than poly(urethane), which allows interaction between molecules in the form of higher hydrogen bonds.
The analysis of the chemical shift in <sup>1</sup>H NMR spectra can be confirmed by the chemical structure of polymers analyzed by FTIR spectroscopy. Based on Figure 10 the proton signals associated with chemical shift values (\(\delta\)) of PU and PUU can be seen, and most proton signals of both polymers have almost the same chemical shift because both polymer molecules were synthesized from the same materials, such as 9-ethoxy-1,10-octadecanediol as a polyol compound and 4,4-methylen-bis phenyl isocyanate (MDI) as a di-isocyanate compound. The difference between both polymers lies in the existence of several signal peaks that appear in the <sup>1</sup>H NMR spectrum of PUU, which are not visible in the <sup>1</sup>H NMR spectrum of PU. In the <sup>1</sup>H NMR spectrum, PUU peaks at the chemical shift were observed at around 5.9 ppm, which are attributed to a — C(O)NH(CH<sub>2</sub>)<sub>2</sub>NH(C(O)– proton and the chemical shift around 2.7 ppm is associated with a -C(O)NH(CH<sub>2</sub>)<sub>2</sub>NH(C(O)- proton. The existence of two signal peaks appearing in the <sup>1</sup>H NMR spectrum of PUU indicates the formation of urea bonds in the PUU, which were formed by interaction between the PU with ethylene diamine as chain extender.
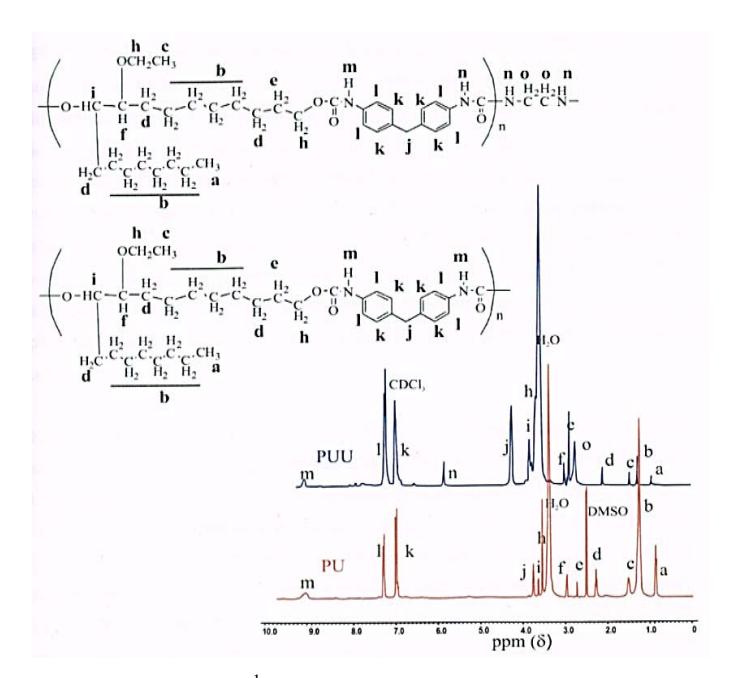
Figure 10 <sup>1</sup>H NMR spectra of PU and PUU.
Based on the analysis of intrinsic viscosity, poly(urethane-urea) has an intrinsic viscosity of 6.44 mL/g, which is higher than that of poly(urethane) (2.19 mL/g). This result indicates that the addition of ethylene diamine as chain extender in poly(urethane) can improve the molecular weight of the polymer to form poly(urethane-urea).
4 Conclusion
Poly(urethane) was successfully prepared by polymerization of diol compounds (9-ethoxy-1,10-octadecanediol) obtained through modification and purification of palm oil and 4,4-methylen-bis phenyl isocyanate (MDI) under nitrogen atmosphere at 85 °C, and poly(urethane-urea) was obtained by addition of ethylene diamine as chain extender into poly(urethane). The 9-ethoxy-1,10 octadecanediol was obtained by several reaction steps, i.e. epoxidation of oleic acid, ring-opening of oleic acid epoxide, and reduction of oleic acid ester. The addition of ethylene diamine as chain extender in poly(urethane) can improve the molecular weight of the polymer to form poly(urethane-urea). This result was supported by an analysis of the chemical structure by FTIR and 1 H NMR spectroscopy. The thermal stability of poly(urethane-urea) is higher than that of poly(urethane) because poly(urethane-urea) has a longer chain than poly(urethane), which allows the interaction between molecules in the form of higher hydrogen bonds.
Acknowledgements
The authors gratefully acknowledge the financial support received from the Research Grant of the Competition Research Program under the Directorate of Higher Education, Department of National Education, the Republic of Indonesia and the Institute for Research – LPPM, Institut Teknologi Bandung (ITB).