
1 Introduction
Heat shock protein 90 (Hsp90) is a molecular chaperone that regulates the maturation, stabilization and activation of many cellular polypeptides important in cell growth, differentiation and survival. Client proteins of Hsp90 include tyrosine kinases, cell cycle regulators, and transcription factors, such as Akt, Raf-1, Her2, and Bcr-Abl [1,2]. In cancerous cells, the client proteins are involved in oncogenic processes ascribed to the ten well-defined hallmarks of cancer; this is confirmed by the fact that Hsp90 expression in the cancerous cells increases 2-3 fold compared to normal cells [3-5].
Hsp90 has three functional conserved domains: the N-terminal domain, which contains an adenosine triphosphate (ATP)-binding site, the middle proteinbinding domain, and the C-terminal domain. The activity of Hsp90 is regulated by the binding and hydrolysis of ATP to ADP (adenosine diphosphate) at the Nterminal domain. Thus, tremendous efforts have recently been performed to find inhibitors capable of binding to the ATP-binding cavity to arrest the function of Hsp90 [6-8].
Hsp90 inhibitors that target the ATP-binding site include natural ansamycin geldanamycin (GA), tanespimycin (17-AAG), and alvespimycin (17-DMAG). However, their development is limited due to chemical structure-related hepatotoxicity [9,10]. The second generation of natural product inhibitors of Hsp90, including Radicicol (RD), exhibits potent anti-tumor properties but lacks chemical stability [11,12]. Structurally different from these inhibitors, protoporphyrin IX (PPIX) was shown by Lee, et al. [13] to effectively inhibit chaperon activity of Hsp90. It was indicated that porphyrin through its pielectron delocalization is necessary for binding to the ATP-binding site of Hsp90. The current study investigated the potentials of several porphyrin derivatives as inhibitors of Hsp90. The anthraquinone group, which was conjugated at the meso-20 position of porphyrin, was expected to interact with hydrophobic residues at the bottom of the Hsp90 binding pocket.
Computational studies are substantially important in the identification of novel inhibitors against a certain protein target in the drug discovery process. While molecular docking is particularly useful for exploring the conceivable orientation of a ligand in the active cavity of the protein target, molecular dynamics simulation is important for evaluating the conformational stability of the drug's target at atomic level [14-16]. In addition, binding free energy calculations using the MM-PBSA method can reveal the pivotal interactions between the ligand recognition and the protein target.
2 Computational Methods
2.1 Macromolecule and Ligand Preparation
The X-ray crystal structure coordinates of Hsp90 co-crystallized with GD were taken from the RCSB Protein Data Bank (PDB: 1YET, resolution: 1.9 Å and Rvalue: 0.193) [17]. The 2D chemical structures of six porphyrin hybrids were designed, i.e. mono-H2PyP-AQ, bis-H2PyP-AQ, tris-H2PyP-AQ, mono-H2PzP-AQ, bis-H2PzP-AQ, tris-H2PzP-AQ (Figure 1), employing GaussView. Geometry optimization of the structures was done by means of the Gaussian 09 program on the 3-21G* basis set at the Hartree-Fock level [18].
\[1. \ R_{1} = R_{3} = H, \ R_{2} = \bigwedge_{N=1}^{Me} : mono-H_{2}PyP-AQ \qquad 4. \ R_{1} = R_{3} = H, \ R_{2} = \bigwedge_{N=N-N-Me}^{Me} : mono-H_{2}PzP-AQ\] \[2. \ R_{2} = H, \ R_{1} = R_{3} = \bigwedge_{N=N-N-Me}^{Me} : bis-H_{2}PyP-AQ \qquad 5. \ R_{2} = H, \ R_{1} = R_{3} = \bigwedge_{N=N-N-Me}^{Me} : bis-H_{2}PzP-AQ\] \[3. \ R_{1} = R_{2} = R_{3} = \bigwedge_{N=N-N-Me}^{Me} : tris-H_{2}PyP-AQ \qquad 6. \ R_{1} = R_{2} = R_{3} = \bigwedge_{N=N-N-Me}^{Me} : tris-H_{2}PzP-AQ\]
Figure 1 The chemical structures of the six porphyrin hybrids.
2.2 Molecular Docking
Molecular docking was carried out with the help of AutoDock 4.2 accessed through AutoDockTools 1.5.6 [19,20]. Kollman charges were assigned to the Hsp90 protein. The size of the grid at the binding pocket of the co-crystal ligand of Hsp90 was set at \(62 \times 62 \times 62\) in the x, y, and z directions, respectively, with grid spacing at 0.375 Å. The Lamarckian genetic algorithm was employed with population size 150 and maximum number of energy evaluations at 2,500,000. One hundred independent docking runs were performed for each porphyrin hybrid, including GD. The default values were used for the other parameters.
2.3 Molecular Dynamics Simulation
MD simulation was performed on the best docking pose of each ligand in complex with Hsp90 using the AMBER16 software package [21,22]. The protein and ligand were prepared using ff14SB and GAFF force fields, respectively [23,24], while partial atomic charges for the ligand were computed using the AM1-BCC method [25]. All systems were then electro-neutralized by applying counter ions, while solvation was performing using explicit TIP3P solvent box with protein-ligand atoms located 10 Å toward the periphery of the box.
Energy minimization was first carried out with restrained protein and ligand \((k = 500 \text{ kcal/mol}\text{Å}^2)\) by using 500 steps of the steepest descent algorithm and 3,500 steps of the conjugate gradient algorithm. A second minimization was performed with the backbone atoms of the protein restrained under the same energy minimization conditions. Final energy minimization was carried out with settings similar to the settings used in the previous minimization but without restraints.
The whole system was then heated in an NVT ensemble from 0 to 300 K in 100 ps with a time step of 0.5 fs and restrained backbone atoms of Hsp90 (\(k = 5 \text{ kcal/mol}\text{Å}^2\)). The system was then equilibrated at 300 K during two subsequent 50 ps with k = 3 and \(k = 1 \text{ kcal/mol}\text{Å}^2\), respectively, which was followed by the final equilibration for 200 ps without restraints. The production step of the whole system was performed for 40 ns with the PMEMD program [22] in an isothermic-isobaric ensemble at 300 K and 1 atm.
In all MD steps, bonds involving hydrogen atoms were constrained with the SHAKE algorithm [26] using a 2-fs time step. The long-range Coulombic interactions were treated with the particle-mesh Ewald method [27] with cutoff at 9.0 Å, while Langevin thermostat collision frequency was employed at 1 ps<sup>-1</sup>. The coordinate files of the production step were saved every 1 ps. The trajectories of each MD run were analyzed using the CPPTRAJ module [28] and the Visual Molecular Dynamics software application [29].
2.4 MM-PBSA Calculations
Prediction of the free energy of binding (\(\Delta G_{\rm bind}\)) was performed by using the Molecular Mechanics-Poisson-Boltzmann Surface Area (MM-PBSA) method on 200 snapshots extracted from 20 to 40 ns of each MD trajectory, from which water molecules and Na ions were removed [30,31]. \(\Delta G_{\rm bind}\) is described through the following Eq. (1) – (3):
\[\Delta G_{\text{bind}} = G_{\text{complex}} - G_{\text{rec}} - G_{\text{ligand}} \tag{1}\]
\[= \Delta E_{MM} + \Delta G_{PB} + \Delta G_{SA} - T\Delta S \tag{2}\]
\[\Delta E_{MM} = \Delta E_{bond} + \Delta E_{angle} + \Delta E_{torsion} + \Delta E_{vdw} + \Delta E_{EEL}\] (3)
where \(G_{\rm complex}\), \(G_{\rm rec}\), and \(G_{\rm ligand}\) are the free energies of the complex, receptor, and ligand, respectively. \(\Delta E_{\rm MM}\) is the gas-phase interaction energy, which comprises bond, angle, torsion, van der Waals and electrostatic energies. \(\Delta G_{\rm PB}\) are the polar solvent free energies calculated by solving the PB equation with a grid size of 0.5 Å. \(\Delta G_{\rm SA}\) are the nonpolar solvent free energies calculated by using the solvent accessible surface area (SASA) with solvent-probe radius set at 1.4 Å. \(T\Delta S\) is the conformational entropy change at temperature T calculated by normal mode analysis.
3 Results and Discussion
3.1 Molecular Docking
The current in silico study aimed to elucidate the inhibitory potential of porphyrin anthraquinone hybrids against Hsp90 at the ATP-binding pocket. The ATP-binding site of the N-terminal domain has a 12 Å diameter width near its entrance, which is wide enough to bind the porphyrin ring (the ring size of the porphyrin being 8-9 Å) and consists of polar Lys58 and Lys112 residues [13]. The binding pocket becomes increasingly hydrophobic toward the bottom, which is composed of Leu107, Gly135, Val136, Gly137, in addition to hydrophilic residue Asp93 [13,17]. The docking study was first performed on the co-crystallized ligand (GD) to validate the docking protocol by measuring the root mean square deviation (RMSD) of the structures of the co-crystallized ligand and the redocked ligand. The binding conformation of GD was consistent with a crystal structure in which hydrogen bonds are formed with Phe138 (Figure 2(a)). The docking result showed that both structures have an RMSD of 0.935 Å, indicating that the docking protocol was reliable (Figure 2(b)).
Figure 2 (a) The docked conformation of GD, (b) Overlying experimental (green) and docked (blue) structures of GD with an RMSD of 0.935 Å.
The docked pose for each cationic porphyrin-anthraquinone hybrid was selected based on the important interactions and docking scores. The cationic porphyrinanthraquinone hybrids containing porphyrin moiety, which has considerable pielectron delocalization, are suited to interact with Lys58 or Lys112. Meanwhile, hydrophobic residues at the bottom of the cavity, such as Phe138, were plotted to interact with the anthraquinone and linker groups. The molecular docking conformation showed that each of the six porphyrin compounds docked into the active site of Hsp90 fitted well into the binding pocket of Hsp90. As expected, each oxygen atom linking porphyrin and anthraquinone groups exhibited hydrogen bonds with hydrophobic residue Phe138, which was also the case for GD. In each porphyrin hybrid interaction, the additional hydrogen bonds, which were not detected in GD interaction, were established between Asp54 and the central hydrogen atoms of porphyrin. The pi-cationic interactions with Lys58 were also established in each porphyrin hybrid; this was absent in the binding of GD. Figure 3 shows the docking poses of bis-H2PyP-AQ, bis-H2PzP-AQ, mono-H2PyP-AQ, mono-H2PzP-AQ, tis-H2PyP-AQ, and tris-H2PzP-AQ in the binding pocket of Hsp90.
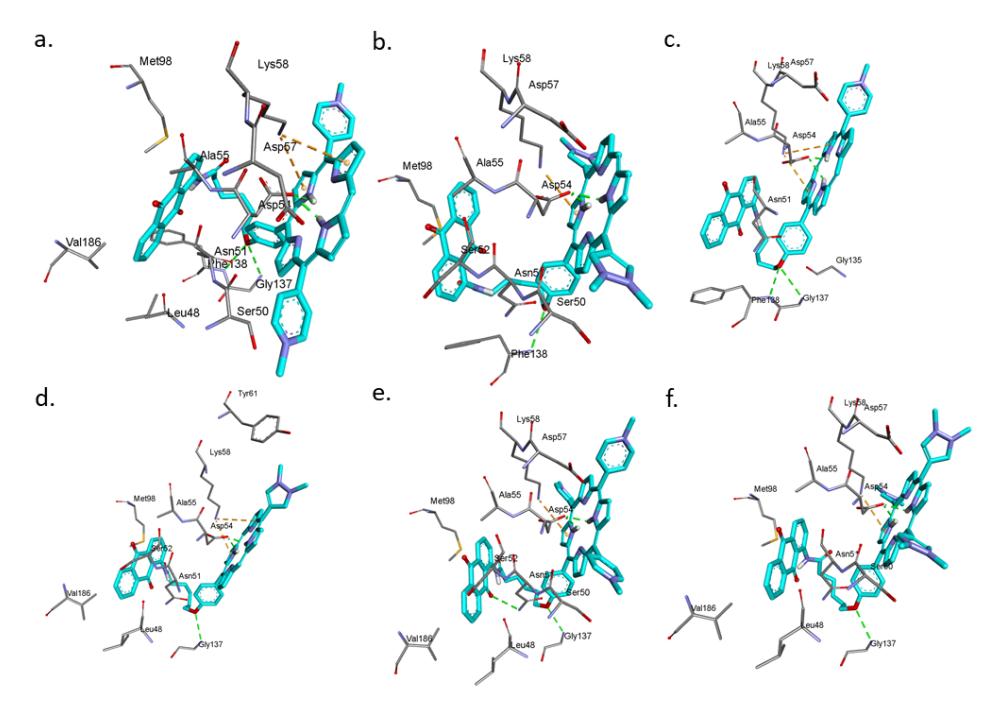
Figure 3 The docking poses of (a) bis-H2PyP-AQ, (b) bis-H2PzP-AQ, (c) mono-H2PyP-AQ, (d) mono-H2PzP-AQ, (e) tris-H2PyP-AQ, and (f) tris-H2PzP-AQ in the Hsp90 binding pocket. The green and orange dashed lines represent hydrogen bonds and pi-cationic interactions, respectively.
3.2 Molecular Dynamics Simulation
Each top-ranked conformation ligand, including GD, obtained from molecular docking was used in a 40-ns MD simulation to further examine the stability of the binding modes of ligands with Hsp90. In the molecular dynamics
simulation, the numbering of residues of Hsp90 from 11 to 223 was changed to 1 to 213. Each system reached equilibrium and there were no significant structural fluctuations during the dynamics run as indicated by the low RMSD of the backbone atoms of the protein (Figure 4).
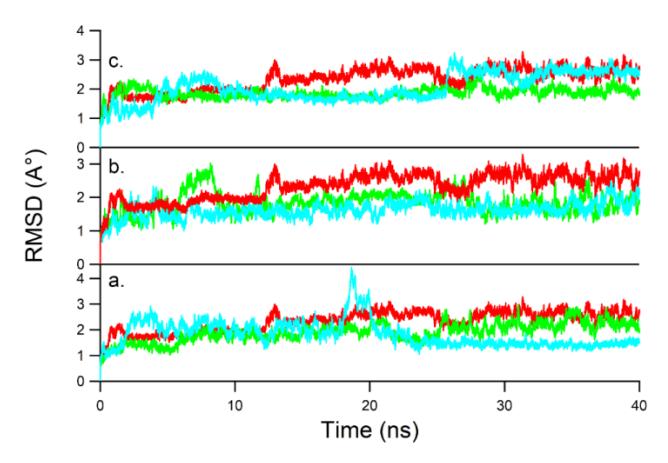
Figure 4 RMSD plot of Hsp90 backbone atoms for: (a) mono-H2PyP-AQ (green), mono-H2PzP-AQ (blue), GD (red); (b) bis-H2PyP-AQ (green), bis-H2PzP-AQ (blue), GD (red); (c) tris-H2PyP-AQ (green), tris-H2PzP-AQ (blue), and GD (red).
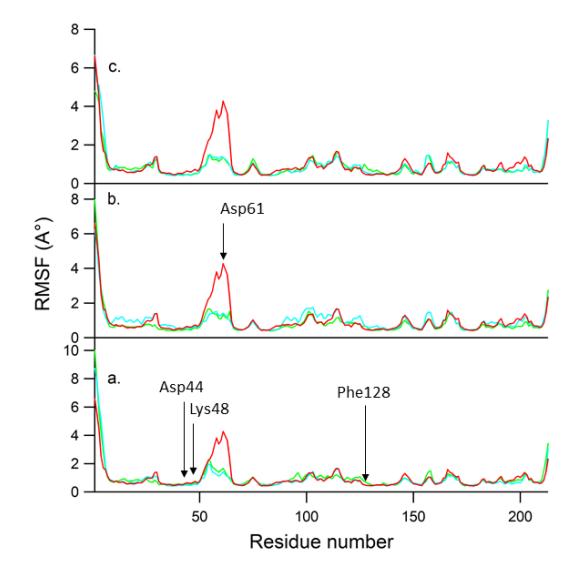
Figure 5 RMSF values of the Cα atoms of each residue: (a) mono-H2PyP-AQ (green), mono-H2PzP-AQ (blue), GD (red); (b) bis-H2PyP-AQ (green), bis-H2PzP-AQ (blue), GD (red); (c) tris-H2PyP-AQ (green), tris-H2PzP-AQ (blue), GD (red).
The residue fluctuation of Hsp90 due to ligand binding was examined by analyzing the root-mean-square fluctuation (RMSF) of all residues. Figure 5 depicts the RMSF profiles, showing that each system had a similar RMSF pattern due to their similar binding modes. High-fluctuation residues were recorded at those located at the N terminal, the C terminal, as well as the loop region (Asp71). Meanwhile, the residues that were involved in the interaction with ligands, including Asp54, Lys58, and Phe138, exhibited high rigidity.
3.3 Prediction of Binding Free Energies
The free energies of binding of porphyrin hybrids and GD to Hsp90 are depicted in Table 1. It was shown that the calculated free energy of binding GD (ΔGpred = −40.46 kcal/mol) was almost four times more negative than that of the observed binding free energy (ΔGexp = −10.61 kcal/mol, which was converted from an IC50 value of 20 nM) [32]. In this study, the entropy term was not calculated and along with the over-predictive tendency of the MM-PBSA calculation, may contribute to the free binding energy discrepancy [25,33]. Furthermore, mono−H2PzP−AQ, bis−H2PzP−AQ, tris−H2PyP−AQ, and tris−H2PzP−AQ exhibited a more negative ΔEPBTOT (−44.48 to −57.01 kcal/mol) compared with ΔEPBTOT of GD (−40.46 kcal/mol), which indicates stronger binding of the porphyrin hybrids than GD. In addition, the porphyrin binding was slightly modulated by the number of meso substituents on the porphyrin core (for comparison, ΔGtris-H2PzP-AQ = −57.01 kcal/mol, while ΔGmono-H2PzP-AQ = −44.48 kcal/mol). In the meantime, the pyrazole group appended to porphyrin macrocycle contributed positively to the binding affinity compared with the pyridine group [25].
| Ligand | ΔEVDW | ΔEELE | ΔEPBCAL | ΔEPBSUR | ΔEPBELE | ΔEPBTOT |
|---|---|---|---|---|---|---|
| mono−H2PyP−AQ | −63.19 | −126.08 | 172.65 | −6.31 | 46.57 | −25.01 |
| mono−H2PzP−AQ | −68.15 | −176.82 | 216.18 | −6.66 | 39.36 | −44.48 |
| bis−H2PyP−AQ | −67.43 | −288.27 | 336.73 | −6.54 | 48.46 | −25.50 |
| bis−H2PzP−AQ | −69.18 | −337.04 | 371.94 | −6.50 | 34.90 | −53.95 |
| tris−H2PyP−AQ | −77.71 | −476.81 | 521.47 | −7.51 | 44.66 | −53.02 |
| tris−H2PzP−AQ | −75.73 | −454.91 | 497.88 | −7.34 | 42.97 | −57.01 |
| GD | −49.68 | −38.38 | 64.61 | −5.06 | 26.23 | −40.46 |
Table 1 The free energy of binding of each ligand to Hsp90.
The electrostatic energies (ΔEELE) and the van der Waals energies (ΔEVDW) of the porphyrin hybrids were favorable for ligand binding and became more negative with the increasing number of peripheral substituents. The nonpolar desolvation energy (ΔEPBSUR) slightly favored porphyrin compounds. However, the total electrostatic terms (ΔEPBELE) of the porphyrin hybrids were higher than those of GD and were highly unfavorable due to unfavorable contribution of the polar desolvation energy (ΔEPBCAL). The MM-PBSA prediction showed that both the electrostatic energies and the van der Waals energies are important in the recognition of porphyrin compounds to Hsp90.
4 Conclusion
The present study demonstrated the binding modes of porphyrin hybrids to Hsp90. All porphyrin hybrids bind to the Hsp90 with similar binding modes and were stable for 40 ns. The porphyrin binding is stabilized by the van der Waals and electrostatic interactions as revealed by MM-PBSA prediction. The binding of porphyrin hybrids, particularly tris−H2PyP−AQ, tris−H2PzP−AQ, bis−H2PzP−AQ, and mono−H2PzP−AQ, are energetically favorable and are stronger than those of the known inhibitor, GD. In addition, the variation of the number of peripheral substituents slightly affects the total binding free energies. It may be useful to further investigate porphyrin hybrids as distinctive scaffolds of Hsp90 inhibitors.
Acknowledgements
The authors wish to thank the Ministry of Research, Technology, and Higher Education of the Republic Indonesia for Hibah Penelitian Pasca Doktor 2017.