
1 Introduction
Urea derivatives, as well-known compounds in the chemical industry, agriculture and medicine, are widely used, especially in the pharmaceutical industry as anticancer drugs. For instance, bromisoval is a urea derivative used as central nervous system depressant and 1-(2-chloroethyl)-1-nitroso-3-propyl urea as well as 1-hydroxyurea serve as anticancer agents [1]. Figure 1 displays the structures of hydroxy urea and 1-(2-chloroethyl)-1-nitroso-3-propylurea in which urea is the mother compound. For this reason, urea acts as
Received January 5<sup>th</sup>, 2019, Revised March 25<sup>th</sup>, 2020, Accepted for publication April 1<sup>st</sup>, 2020. Copyright © 2020 Published by ITB Institute for Research and Community Services, ISSN: 2337-5760, DOI: 10.5614/j.math.fund.sci.2020.52.2.3
pharmacophore. However, due to its strong hydrophilicity, urea has an unsatisfactory level of biological membrane penetration. This results in insufficiently optimized pharmacological activities. Hydroxyurea, for instance, is widely used as an anticancer drug despite the fact that it does not exhibit the desired outcome. With this in mind, the modification of urea structures to produce novel urea derivatives with stronger lipophilic properties is important. Thus, membrane penetration and pharmacological activities will eventually improve.
\[H_2N\] \(H_2\) \(H_2N\) \(H_2\) \(H_2N\) \(H_3\) \(H_4\) \(H_5\) \(H_5\) \(H_5\) \(H_6\) \(H_7\) \(H_8\) \(H_8\) \(H_8\) \(H_8\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H_9\) \(H\)
Figure 1 The structure of urea and derivatives.
In the coming years, the number of cancer patients will increase, possibly to about 14 million of cancer cases per year [2]. In response to urea derivatives continually being developed, several researchers have conducted modifications to the structure of urea. Hardjono, et al. [3]synthesized 1-(benzoyloxy) urea compounds that showed anticancer activity on HeLa cell lines. The compound and derivatives of benzoyl-N, N'-diethylurea were synthesized by Diyah, et al. [4], which displayed anticancer activity against MCF7 cell lines [4]. N- (allylcarbamothioyl)benzamide was synthesized by Widiandani, et al. [5] and showed a cytotoxic effect on human breast cancer cell lines (T47D). The study by Kesuma, et al. [6] successfully synthesized N-(phenylcarbamothioyl) benzamide compound that exhibited anticancer activity against MCF-7 cell lines. Overall, any changes made in the urea structure of derivatives will also alter its physicochemical properties, such as hydrophobicity, electronic and steric effects, and pharmacological activity [7].
Given that a growing number of patients will be diagnosed with cancer over time, it is crucial to carry out further studies to obtain alternative urea derivates as anticancer drugs. Phenylurea is one example. Phenylurea is a urea derivative compound that has one phenyl group, which binds the nitrogen atom to free amino groups, making the urea more hydrophobic. This enables easier biological membrane penetration and eventually leads to stronger pharmacological activity compared to hydroxyurea. Some researches have been conducted to exploreN-phenylurea derivatives as anticancer agents. Song, et al. [8] synthesized derivatives of 3-haloacylamino phenylureas with antitumor potency. A derivative with bromoacetyl substituent bound at the N' position, had anticancer potency on 8 human tumor cell lines with an IC50 of 0.38-4.07 μM. Eldehna, et al. [9] synthesized 1-(2-methyl-6-arylpyridine-3-yl)-3 phenylureas and found that the 5l derivative had a potent anticancer effect on lung cancer cells A549 and colon cancer HCT-116, with an IC50 of 3.22 ± 0.2 and 2.71 ± 0.16 μM. A series of 4-substituted-N-(phenylcarbamoyl)- 3pyridinesulfonamides was synthesized by Szafranski and Slawinsk [10]. The compounds, namely N-(4-chlorophenyl) carbamoyl)-4(4-(3,4-dichlorophenyl) piperazine-1-yl) pyridine-3sulfonamide had antitumor potency against leukemia, colon cancer and melanoma with an IC50 of 13.6-14.9 μM.
Developing another N-phenylurea derivative as a new anticancer compound requires molecular design. The Molegro Virtual Docker program used in this study predicts the existence of synchronized interactions with its receptors, which make the anticancer pharmacological activities more potent. Checkpoint kinase 1 (CHK1) in this research was used as the receptor target from the Nphenylurea. CHK1 is a human nuclearserine/threonine-protein kinase. Upon DNA damage, CHK1 is activated and can phosphorylate, which is necessary for S and G2 arrest. Inhibition of CHK1 allows thedamaged DNA cells to progress prematurely into mitosis, resulting in apoptosis. Therefore, abrogation of the S and G2 checkpoints should lead to an increased and selective sensitivity of cancer cells to DNA damage in p53-deficient cells, so that selective inhibitors of CHK1 may be of great therapeutic value in cancer treatment [11].
The N-phenylurea derivative that was synthesized in this study is N- (phenylcarbamoyl)benzamide. After going through the docking process, the rerank score was obtained and compared with that of inhydroxyurea. This is the standard method to predict the pharmacological activity of a derivative as a new anticancer agent. [12]. In order to obtain predictions of absorption, distribution, metabolism, excretion and the toxicity of N-phenylurea as a new derivative compound, ADMET Predictor was used. This was to ensure that the predictions of the data were available when it was being developed as an anticancer drug candidate. Changes in ADMET Predictor's predictions after the new phenylurea derivative had been synthesized would certainly occur and they would be different from those for the mother compounds [13].
2 Material and Methods
2.1 Solvents and the Other
The compounds and solvents for synthesis and physicochemical analysis used in this study were: N-phenylurea, benzoyl chloride (Sigma Aldrich), pyridine, acetone, ethyl acetate, n-hexane, chloroform, ethanol, and methanol (p.a. quality), Kieselgel 60 F254, DMSO-d6 (E.Merck).The componentsused in the cytotoxic activity test were: N-(phenylcarbamoyl) benzamide, HeLa cell line culture, DMEM culture, DMSO, phosphate buffer saline (PBS), MTT [3-(4,5 dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide], SDS 10% in 0,1 N HCl, and standard compound hydroxyurea (Sigma Aldrich).
2.2 Instruments
The instruments used for synthesis and structural analysis were: a UV-Vis Shimadzu HP 8452 Aspectrophotometer, a Jasco FT-IR 5300 spectrophotometer, an NMR Hitachi R-1900-90 MHz spectrometer, a JEOL-JMS 600 mass spectrometer, an Electrothermal Mel-Temp, a Corning hot plate P 351, an Shimadzu LM-20 analytical balance. The instruments used for the cytotoxic activity test were: a micropipette with a200-1000 μL tip, test tubes, microplates, conical tubes, and ELISA Reader.
2.3 Procedures
2.3.1 Molecular Docking
Hardware and software used: Lenovo computer; operating system Windows 10, 64-bit; Intel Core i5-7200 U; CPU@250 GHz, 8.00 GB RAM; Chem Bio Draw Ultra, version 12 (Cambridge Soft); Molegro Virtual Docker version 5.5 (CLC bio). The in silico process was done by molecular docking in the initial stage using the ChemBioDraw Ultra program, version 12.0. The 2D structure of the N-(phenylcarbamoyl)benzamide compound was analyzed and determined. In the next step, the compound was designed with a 3D model and the most stable design was selected using ChemBio 3D Ultra, version 12.0. After that, finding the crystal structure of checkpoint kinase 1 (CHK1) enzyme, ID PDB: 2YWP with a ligand 1-(5-chloro-2,4-dimethoxyphenyl)-3-(5-cyanopyrazin-2-yl)urea (A42_1) and alignment between the active compounds N-(phenylcarbamoyl) benzamide and the CHK1 enzyme was carried out. A 3D depiction of the harmony between the active compound N-(phenylcarbamoyl)benzamide and the receptor was needed to determine the presence of the active compound in the appropriate cavity. The next step was to verify the binding of the active compound with several amino acids associated with the hydrogen bonds and the steric interactions. The final stage was involved docking the compound using Molegro Virtual Docker, version 5.5 until the rerank scores (RS) were obtained [11,13,14].
2.3.2 Molecular Dynamics Simulation
In order to validate the calculation resulted from the molecular docking procedure, a molecular dynamics simulation was applied [15]. This procedure was performed using the Amber Molecular Dynamics package program for a 10-ns simulation [16]. A post-processing procedure was applied to calculate the binding free energy between the receptor and each ligand. The binding free energy calculation was carried out using the MMPBSA.py module, which is part of the Amber Molecular Dynamics software package [17].
The receptor–native ligand complex model (PDB ID 2YWP) was downloaded from the PDB server. This complex was pre-processed using the UCSF Chimera program [18]. Atomic partial charge was added to each atom of the receptor using the molecular mechanics (MM) method with the AMBER ff14SB force field and to each atom of the ligand using the semi-empirical method AM1-BCC [19]. The other ligands in this experiment (ndp and 1hd) were converted from a 2D structure to a 3D structure using semi empirical method PM6.
2.3.3 ADMET Prediction
Hardware and software used: Lenovo computer; operating system Windows 10, 64-bit;Intel Core i5-7200 U; CPU@250 GHz, 8.00 GB RAM; Chem Bio Draw Ultra, version 12 (Cambridge Soft); Online SMILES Translator; and pkCSM online tool. Pharmacokinetic properties prediction (ADMET: absorption, distribution, metabolism, excretion, and toxicity) from N-(phenylcarbamoyl) benzamide was carried out using the pkCSM online tool. The compound was drawn as a 2D molecular structure and then the structure was copied to the ChemBio 3D Ultra program to create a 3D structure, stored in *.sdf files. Afterwards, the compound was translated into the SMILE format using the Online SMILES Translator (http://cactus.nci.nih.gov/translate/). In the SMILE format, the compound was processed using the pkCSM online tool (htpp://biosig.unimelb.edu.au/pkCSM/prediction) to predict the ADMET of the compounds [20,21].
2.3.4 Synthesis Procedure
In a 200-ml round bottom flask, 0.03 moles of N-phenylurea were mixed with 40 ml of tetrahydrofuran and 4 ml of pyridine. At 5 °C, a 0.01-mole benzoyl chloride solution was added little by a little to 20 ml of tetrahydrofuran while stirring with a magnetic stirrer. After that the mixture was refluxed and stirred for 6 hours, the reaction was terminated and tetrahydrofuran was evaporated in the rotary evaporator. A saturated sodium bicarbonate solution was added while stirring until no air bubbles (froth) appeared. The product was filtered with a Buchner funnel and the solid was washed with 50 ml of water 2 times and subsequently with 10 ml of ethanol 2 times. Recrystallization obtained by dissolving the solid with enough ethanol while stirring on a heater (hot plate). The solution was filtered in a hot state, the filtrate was left at room temperature to cool overnight. The formed crystals were then filtered with a Buchner funnel and washed with 10 ml of ethanol 2 times. Depending on the synthesized compound obtained, recrystallization can be carried out with other suitable solvents, such as acetone-water. The formed crystals were moved to a petri dish, then dried in an oven at 50 ºC until constant weight gained. [21,22]. The synthesis reaction can be seen in Figure 2. Structure identification of the N- (phenylcarbamoyl)benzamide was done by using a UV-Vis and IR spectrophotometer, 1H-NMR, andmMass spectrometer [23].
N-phenylurea Benzoyl chloride N-(phenylcarbamoyl)benzamide
Figure 2 The synthesis reaction of N-(phenylcarbamoyl)benzamide.
2.3.5 The Cytotoxic Activity Test
To evaluate the cytotoxic effect of the compound, an MTT [3-(4,5 dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay was performed. The test solution was prepared from N-(phenylcarbamoyl) benzamide compound at 5000 mg/mL using DMSO solvent. Next, a series of standard solutions of 250, 500, 750, 1000 and 2000 mg/mL were prepared. In addition, the standard solution of hydroxyurea was prepared with the same concentration as positive control with solvent blank as negative control.
Cancer cells/HeLa cells and normal cells were prepared in the form of suspension cells with a density of 1.05-2.106 and added into microplates of 100 ml each. Media control was also prepared at 0.2 mL from each solution (test compound, standard compound, and negative control) and inserted in a microplate, which was incubated afterwards in 5% CO2 incubator for 24 hours at 37 °C and pH 7.4-7.7.
MTT solution was prepared by diluting 1 mL of MTT stock, 50 mg of MTT in 10 ml PBS so that 0.5 mg/mLof product was obtained for the treatment test. After incubation, the cell medium was discarded and the cells were washed with PBS. 100 mL of MTT reagent was added to each plate after which the product was incubated for 24 hours in a CO2 incubator. After formazan was formed, 100 mL of SDS 10% was added to the 0.01 N HCl. The plate was wrapped in aluminum foil and incubated in a dark place at room temperature for 24 hours. The next stage was observation of absorbance of each well using Eliza Reader at a wavelength of 595 nm. The absorbance was higher when the cancer cells/normal cells were alive. Calculation of IC50 was done by probit regression analysis using SPSS [13,20].
3 Result
3.1 Molecular Docking
The amino acid types and numbers bound to the compound are listed in Table 1 and Figure 3.
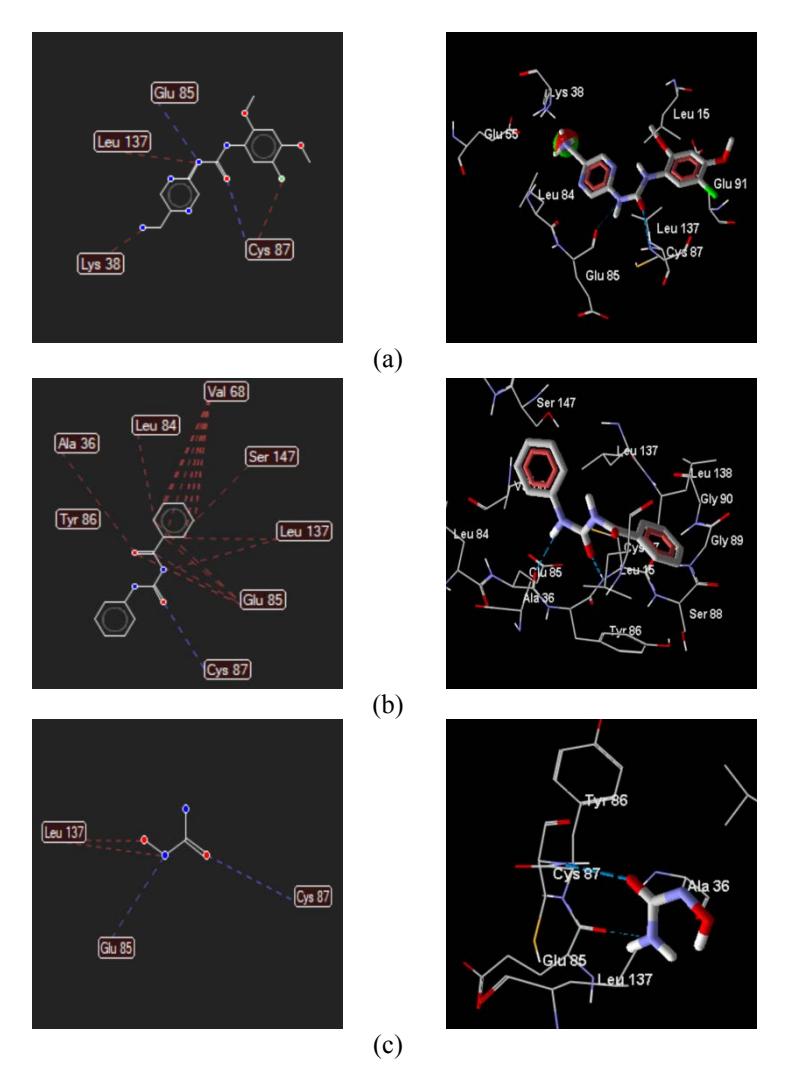
Figure 3 Interaction of the compounds with the amino acid: (a) ligand: A42_1, (b) N-(phenylcarbamoyl)benzamide, and (c) hydroxyurea.Blue lines indicate hydrogen bonds, red lines indicate steric interaction.
Table 1 Rerank Score (RS) and amino acid interaction of the N- (phenylcarbamoyl)benzamide (PCB) compound, the hydroxyurea (HU) and the ligand (A42_1).
| No | Compounds | Rerank score (kcal/mol) | Amino acid interaction | |
|---|---|---|---|---|
| Steric interaction | Hydrogen bond | |||
| 1 | PCB | -72.0603 | Ala36; Leu 84; Val 68; Ser 147; Leu 137; Glu 85. | Cys 87 |
| 2 | HU | -32.1514 | Leu 137 | Glu 85; Cys 87 |
| 3 | A42_1 | -91.8919 | Lys 38; Leu 137; Cys 87 | Glu 85; Cys 87 |
3.2 Molecular Dynamics Simulation
The binding free energy calculated from this simulation is listed in Table 2.
Table 2 Binding free energy between receptor and ligand after 10-ns simulation.
| Energy | Ligand | |||
|---|---|---|---|---|
| Component | A42_1 | PCB | HU | |
| VDWAALS | -36.9265 ± 0.1477 | -19.0250 ± 3.6622 | -0.2275 ± 0.9118 | |
| EGB | 6.6578 ± 0.0447 | 3.6743 ± 0.5790 | 0.1129 ± 0.3289 | |
| ESURF | -4.7948 ± 0.0213 | -2.5733 ± 0.4644 | -0.0406 ± 0.1912 | |
| DELTA G gas | -36.9265 ± 0.1477 | -19.0250 ± 3.6622 | -0.2275 ± 0.9118 | |
| DELTA G solvent | 1.8630 ± 0.0345 | 1.1010 ± 0.4396 | 0.0722 ± 0.1568 | |
| DELTA TOTAL | -35.0636 ± 0.1441 | -17.9240 ± 3.7167 | -0.1553 ± 0.7907 | |
3.3 ADMET Prediction
The ADMET prediction of the N-(phenylcarbamoyl) benzamide compound from the pkCSM online tool is shown in Table 3. On the basis of the chemical contents above it can be concluded that N-(phenylcarbamoyl)benzamide compounds were successfully synthesized in this study.
Table 3 ADMET prediction of the N-(phenylcarbamoyl)benzamide compound.
| Absorption | Intestinal absorption (human) in % | 91.436 |
|---|---|---|
| Skin permeability (log Kp, cm/h) | - 0.37 | |
| VDss (logL/kg) | - 0.182 | |
| Distribution | BBB permeability (logBB) | 0.115 |
| CYP2D6 substrate | No | |
| Metabolism | CYP2D6 inhibitor | No |
| Renal OCT2 substrate | No | |
| Excretion | Total clearance (log mL/min/kg) | 0.263 |
| Ames toxicity | No | |
| Toxicity | LD50 (mol/kg BW) in rat | 1.901 |
| Hepatotoxic | No |
3.4 Chemistry of the N-(phenylcarbamoyl)benzamide
N-(phenylcarbamoyl)benzamide, yield: 82%; MP: 195°C; UV(methanol, \(\lambda\)max, nm): 204, 232, 272 (sh); FT-IR (KBr pellet; cm<sup>-1</sup>): 3240 (NH sec), 1698 and 1676 (2 C=O ureide), 1600, 1475 (C=C aromatic); <sup>1-</sup>H-NMR (DMSO-d<sub>6</sub>, σ, ppm): 7.00-8.10 (m, 10H, 2 C<sub>6</sub>H<sub>5</sub>), 10.60 (s, 1H, NH), 11.20 (s, 1H, NH); MS (EI; m/e), 240 (M)<sup>+</sup>.
3.5 Cytotoxic Activity Test
The viability (%) of the HeLa cell lines after treatment by the compounds is shown in Figure 4.
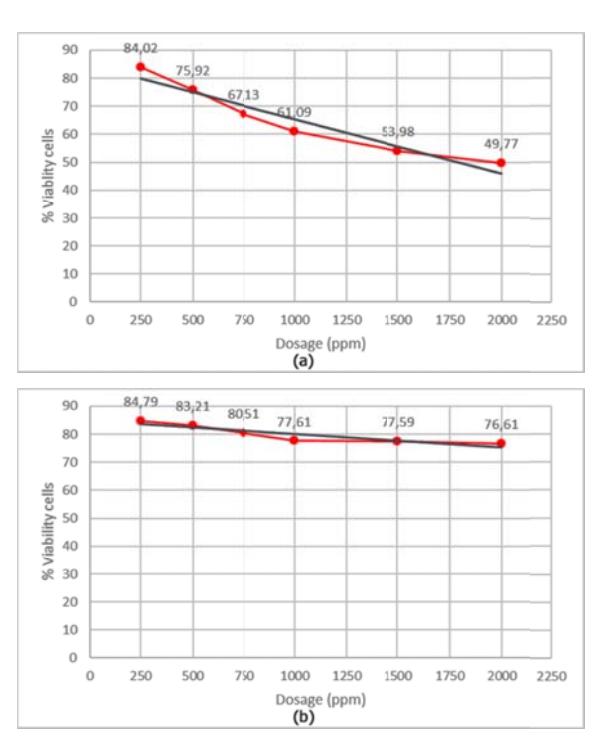
Figure 4 The viability (%) of the HeLa cell lines after treatment by: (a) the N-(phenylcarbamoyl)benzamide compound, and (b) the hydroxyurea.
In Figure 4 it can be seen that the viability (%) of the HeLa cell lines decreased significantly after being treated with N-(phenylcarbamoyl)benzamide compound (a) compared to hydroxyurea (b). This means that the N-(phenylcarbamoyl) benzamide compound had a higher cytotoxic effect than hydroxyurea. According to the probit analysis carried out using the SPSS program, the IC<sub>80</sub> of the N-(phenylcarbamoyl)benzamide compound was 191.1 ppm or 0.8 mM while the IC<sub>80</sub> of the hydroxyurea as standard compound was 1036.3 ppm or 4.3 mM.
4 Discussion
4.1 Molecular Docking and Molecular Dynamics Simulation
The activity of the drug was predicted by molecular docking using the value of RS as the indicator. Molecular docking is a prediction to describe bindings among active compounds, the standard compound and the ligand with its receptors hypothetically through in silico studies. The RS value is a hypothetical presentation of a compound's binding affinity with receptors described as energy binding. If the RS of the active compound is smaller compared to that of the standard compound, then the binding energy to the receptors becomes more stable. Thus, it can be predicted that the biological activity of the active compound is improving compared to the standard compound.
The prediction of anticancer activity from the N-(phenylcarbamoyl)benzamide conducted by molecular docking had an RS value of -72.0603 kcal/mol, i.e. greater than that of hydroxyurea (-32.1514 kcal/mol). Yet, this is still smaller than the RS value of ligand A42_1 (Table 1). A smaller RS value indicates that the bonds between drug-receptor interactions are more stable, giving better anticancer activity. Table 2 shows the result of binding free energy between the receptor and its ligand using molecular dynamics simulation. It can be seen that the delta total of free energy in N-(phenylcarbamoyl)benzamide (-17.9240 ± 3.7167) was smaller than that in HU (-0.1553 ± 0.7907). Similarly, the binding energy of N-(phenylcarbamoyl)benzamide compound was also smaller that that of its ligand. This results in a more stable binding between the receptor and the N-(phenylcarbamoyl)benzamide compound when compared to the binding between the receptor and the HU compound. This also strengthens the docking results and leads to higher cytotoxic activity in the N- (phenylcarbamoyl)benzamide compound when compared to the HU compound.
The amino acids types and numbers bound to the compound are listed in Table 1 and Figure 3. From Table 1, it can be seen that all compounds bind Cys 87 by hydrogen bonds and Leu 137 by steric interaction. Both the ligand and the hydroxyurea were bound to Glu 85 by hydrogen bonds, while N- (phenylcarbamoyl)benzamide was bound to Glu 85 by steric interactions. This suggests that the amino acid plays an important role in the anticancer activity. The N-(phenylcarbamoyl)benzamide compound produced a larger number of steric interactions with the amino acid at the 2YWP receptor compared to the hydroxy urea. Based on the binding of the drug and the amino acids it can be predicted that the greater number of hydrogen bonds and steric interactions will result in more stable bonding between the drug and the receptor, thus providing better pharmacological activity as well. The prediction suggests that N-
(phenylcarbamoyl)benzamide compound has better inhibitory activity compared to hydroxyurea. Moreover, the higher logP value of the N- (phenylcarbamoyl)benzamide compound also enables better membrane penetration.
5 ADMET Prediction
Lipinski, et al. [24] analyzed 2,245 drugs from the World Drugs Index and came up with Lipinski's rule of five for orally administered drugs, because all the values are multiples of five. If the compound has a molecular weight greater than 500, the log coefficient partition of octanol/water (logP) is greater than +5, the number of donor H-bonds (HBD), which are expressed by the number of O-H and N-H groups, is greater than 5, and the number of H-receptors (HBA), which are expressed by the number of O and N atoms, is greater than 10. This makes the compound difficult to absorb and its permeability low. The N- (phenylcarbamoyl)benzamide had a molecular weight of 240.26 (less than 500), a log coefficient partition of octanol/water (ClogP) 3.39 (less than 5). The number of OH and NH groups was 2, i.e. not more than 5, while the number of O and N atoms was 4, i.e. less than 10. These findings indicate that N- (phenylcarbamoyl)benzamide compound can be easily absorbed and has a high level of permeability.
The intestine is normally the primary site for absorption of a drug from an orally administered solution. The ADMET Prediction method predicts the proportion of compounds that are absorbed through the human small intestine. A molecule with an absorbance of less than 30% is considered to be poorly absorbed [11]. Imming [25] states that the ideal new drug has oral bioavailability ≥90% with no inter-individual variation. As can be seen in Table 2, the N-(phenylcarbamoyl)benzamide had an intestinal absorption (human) of 91.436%. This means that the compound has good oral bioavailability.
Skin permeability is a widespread interest in the development of transdermal drug delivery. It predicts whether the given compound is likely to be skin permeable, expressed as skin permeability constant logKp (cm/h). A compound is considered to have a relatively low skin permeability if it has logKp > -2.5.As can be seen inTable 2, the N-(phenylcarbamoyl)benzamide had a logKp of -0.37 (cm/h),which means that the compound had a relatively low skin permeability.
The theoretical volume that the total dose of the drug needs to distribute evenly to give the same concentration in the blood is known as the distribution volume (VDss). If a drug has a high VDss value it can be said that is distributed more evenly in the network than in plasma. Pires, et al. [26] state that a compound has a low distribution volume if log VDss <-0.15 and a high distribution volume if log VDss > 0.45. As can be seen in Table 2, the N- (phenylcarbamoyl)benzamide compound had a log VDss of -0.182, i.e. smaller than -0.15, which means that it has a low distribution volume. Pires, et al. also state that at log BB well > 3, a compound is able to penetrate well but cannot be distributed properly if log BB < -1. As can be seen in Table 2, the N- (phenylcarbamoyl)benzamide compound had a log BB value of 0.115, i.e. smaller than 3, so the distribution was low and it could not penetrate the BBB well [26]. In the body, cytochrome P450 is an important detoxification enzyme that is mainly found in the liver. Foreign organic compounds, including drugs, are oxidized and facilitate excretion by this enzyme. It is important to assess the ability of compounds to inhibit cytochrome P450, which is represented by cytochrome P2D6 isoform (CYP2D6) in this study. As can be seen in Table 2, the N-(phenylcarbamoyl)benzamide compound did not affect cytochrome P450.
The compound excretion process can be predicted by measuring the total clearance (Cltot) and the renal organic cation transporter 2 (OCT2) substrate. The combination of hepatic clearance (metabolism in the liver and bile) and renal clearance (renal excretion) is Cltot. In Table 3 it can be seen that the Cltotof the N-(phenylcarbamoyl)benzamide compound was 0.263 log mL/min/kg, which is the predicted rate of excretion of the compound. Organiccation transporter 2 (OCT2) is a transporter in the kidney that plays an important role in the disposition and clearance of drugs and endogenous compounds. In Table 3 it can be seen that the N-(phenylcarbamoyl)benzamide compound did not affect OCT2, so it is not an OCT2 substrate.
The toxicity of the compound can be determined by the Ames toxicity test, which is widely used to access the mutagenic potential of compounds using bacteria. If the test has a positive value it can be said that the compound is mutagenic and can act as a carcinogen. From Table 3 it can be seen that the N- (phenylcarbamoyl)benzamide compound did not have mutagenic potential and that it is not hepatotoxic. The LD50 in rats was 1.901 mol/kg BW, which means that the compound is predicted to have relatively low toxicity, because to kill 50% of animals a dose of 457 mg/kg BW is needed.
6 Conclusion
From the above description, N-(phenylcarbamoyl)benzamide compound was successfully synthesized with higher cytotoxic effect on HeLa cell lines compared to standard hydroxyurea, supporting a higher RS value and good pharmacokinetic properties. The compound was neither mutagenic nor hepatotoxic. Therefore, the N-(phenylcarbamoyl)benzamide can be developed as a candidate anticancer drug.
Acknowledgment
The authors are very grateful to the Faculty of Pharmacy Airlangga University for the funding of this study.
Conflicts of Interest
The authors declare no conflicts of interest.