
1 Introduction
Janus kinases (JAKs) are members of the family of intracellular tyrosine kinases. They are phosphotransferases that bind to cytokine receptors, including interleukins, interferons, and many other cytokines [1]. Janus kinases, along with their signal transducer and activators of transcription proteins (STAT), mediate transduction of signaling cascades through the JAK-STAT-PI3K-MAPK pathway, which is crucial for the differentiation, proliferation and survival of a variety of cell types [2-5].
The JAK family consists of 4 isoforms, namely JAK1, JAK2, JAK3 and Tyk2. They have similar biochemical properties due to high sequence homologies. While JAK1, JAK2, and Tyk2 are predominantly found in a variety of cell types, JAK3 is chiefly expressed in hematopoietic, myeloid and lymphoid cells [6]. More specifically, JAK3 is limited to the gamma chain (γc) subfamily of cytokines in both humans and mice [3,4,7,8]. Thus, inhibition of JAK3 is a promising approach for the elimination of autoimmune and inflammatory diseases [9]. Various JAK3 inhibitors have been researched, including tofacitinib and ruxolitinib [8,9]. However, several adverse effects, including increased risk of infection, lymphoma, cancer and tuberculosis, were noted in the clinical use of these inhibitors, which are attributable to the inhibition of multiple JAKs due to the high extent of structural conservation of the ATP binding site of JAK [1,3,4,10,11]. Thus, the search for more selective and potent JAK3 inhibitors is being actively pursued, both in academic institutions and pharmaceutical companies. Here we report a pharmacophore model of JAK3, which was used to screen the Indonesian Medicinal Plants Database (HerbalDB) [12]. Pharmacophore-based in silico screening was combined with a molecular docking simulation to identify novel potential JAK3 inhibitors from the Indonesian herbal database. Further, a molecular dynamics simulation was performed to confirm the stable interaction between the ligands and JAK3. Finally, the Molecular-Mechanics Poisson Boltzmann Surface Area (MM-PBSA) method was utilized to assess the affinity of the hit molecules.
2 Method
2.1 Structure-based Pharmacophore Modeling
A pharmacophore model was built by employing the structure of a carboxamide (1NX) complexed with JAK3, which was retrieved from Research Collaboratory for Structural Bioinformatics PDB ID 3ZEP [13]. Validation of the pharmacophore model was conducted by performing screening against 100 actives taken from BindingDB [14] and 7500 decoys retrieved from the DUD-E database [15]. This task was completed by using the LigandScout Advanced 4.3 software [16]. The validated model was employed to identify potential inhibitors of JAK3 against the Indonesian Medicinal Plants Database (HerbalDB) [12], which contains 1379 molecules.
2.2 Molecular Docking, Molecular Dynamics and MM-PBSA Energy Calculation
The obtained hit molecules from pharmacophore screening were then subjected to molecular docking simulation to the JAK3 protein. A set of polar hydrogens was added and Kollman charges were assigned to the JAK3 in the receptor
preparation step. This task was performed using AutoDockTools 1.5.6. Each ligand was then docked into an active site of JAK3, which was established based on the grid box, which was the center of the native ligand (1NX) coordinates. The molecular docking was carried out by using the iDock software [17]. Each docked ligand conformation was analyzed and visualized using Discovery Studio Visualizer 2016 and the three molecules with the best binding affinities and with good interaction were subjected to a molecular dynamics simulation.
The molecular dynamics simulation was performed for each four ligands complexed with JAK3 employing the AMBER16 package [18-21]. The leap module was used to prepare each complex using the ff14SB force field [22] for the protein and the GAFF force field [23], and AM1-BCC [24] for the ligands. The protein was protonated according to the default Amber procedure. The minimization, heating and equilibration steps, the MM-PBSA calculation and the parameters employed are described in our previous publication [21]. The production MD simulation was conducted for 50 ns in NPT ensemble [21].
3 Results and Discussion
The structure of a carboxamide (1NX) complexed with JAK3 was adopted for building the pharmacophore model, which resulted in several pharmacophore models. From these models, one hypothesis was found, consisting of one hydrogen bond (H-bond) acceptor, two H-bond donors, and two hydrophobic features. Figure 1 shows the generated pharmacophore model.
Figure 1 3D pharmacophore model, consisting of one H-bond acceptor (red dotted lines), two H-bond donors (green dotted lines), and two hydrophobic (yellow sphere) features.
The pharmacophore model was then validated against 100 actives and 7500 decoys. It was found that the area under curve (AUC) of the receiver operating characteristic (ROC) curve was 0.52. Meanwhile the calculation of the goodness of hit score (GH score) gave a result of 0.76. Both parameters indicated the validity of the pharmacophore model developed. Figure 2 displays the AUC of the ROC curve.
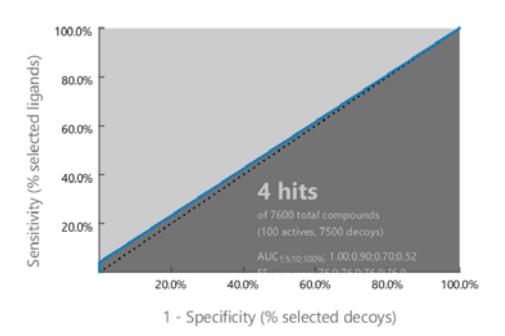
Figure 2 The AUC plot of the ROC curve.
Screening for hit molecules against the Indonesian Herbal Database (HerbalDB) yielded 28 hit molecules. Molecular docking of each hit molecule into JAK3 gave binding energies between −7.52 and −9.97 kcal/mol. These energy values are comparable to the binding energy of a docked native ligand (1NX) of −8.82 kcal/mol. Further, we selected the three best docked hit molecules for an MD simulation, i.e. camelliaside B/Lig2 (E = −9.97 kcal/mol), 3-Ogalloylepicatechin-(4beta-6)-epicatechin-3-O-gallate/lig 10 (E = −9.91 kcal/mol), and mesuaferrone B/Lig17 (E = −9.74 kcal/mol). Figure 3 shows the hit structures of the three best docked hit molecules.
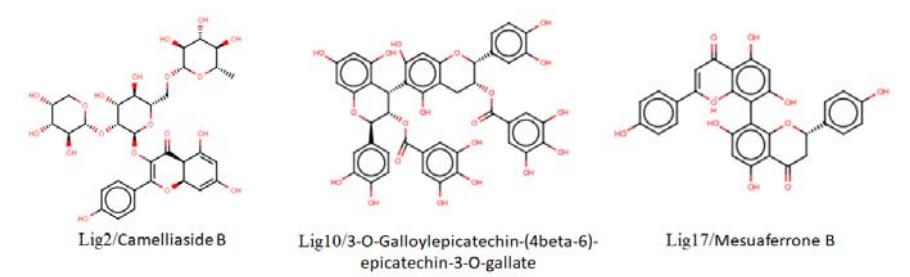
Figure 3 The hit structures of the three best docked hit molecules.
Binding conformations of the hit molecules occurred in the binding site of JAK3. Lig2 formed five H-bond interactions with Lys830, Cys909, Arg911, Asp967, and Arg953, while Lig10 formed H-bond interactions with Lys855, Leu905, Glu903, Lys830, Asp912, Met902, and Gln827. The binding of Lig17 was also corroborated with H-bonds Cys909, Asp967, and Leu905. Amino acid residue Cys909 is known as a key active site residue of JAK3. Figure 4 depicts the binding interactions of each ligand within the binding pocket of JAK3.
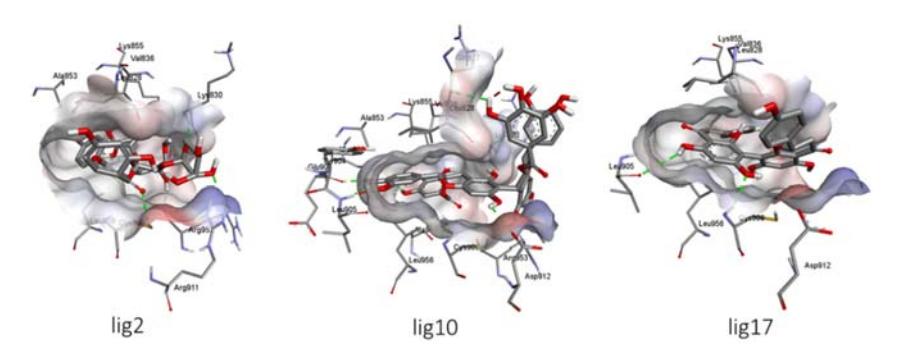
Figure 4 The binding interactions of Lig2, Lig10 and Lig17 within the binding pocket of JAK3.
3.1 Molecular Dynamics Simulations
As the molecular docking method does not consider the physiological condition for stable interaction of the hit molecules, each three best docked hit molecules complexed with JAK3 were subjected to an MD simulation. The MD simulation was carried out for 50 ns to see whether the ligands would retain interaction with JAK3. The values of root mean square deviation (RMSD) during the MD simulation run were used to monitor the stability of the complex. Figure 5 shows the RMSD values of the heavy atoms (Cα, C, N, O) of JAK3.
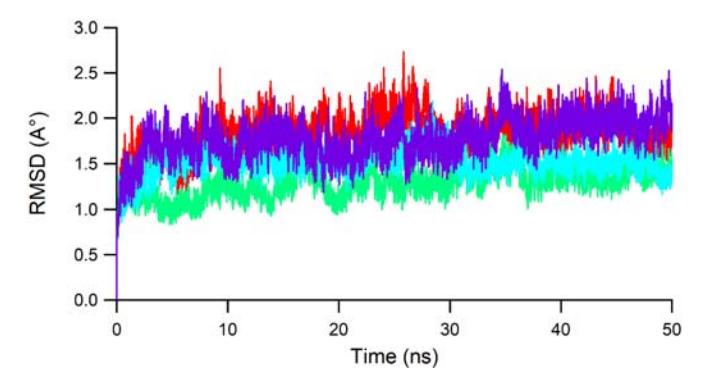
Figure 5 The RMSD values of the main protein atoms of each JAK3 ligand system during 50 ns MD accounted for Lig2, Lig10, Lig17, and 1NX, colored red, green, blue, and purple, respectively.
It is noted that each ligand maintained stable interaction with JAK3. Lig10 (green) and Lig17 (blue) had lower RMSD values than 1NX, which indicates more stable interactions. All fluctuations of the RMSD values for all ligands were under 3 Å, which can be considered stable conformational changes, during 50 ns.
The fluctuation of the individual amino acid residues during the MD simulation was recorded as a root mean square fluctuation (RMSF) plot. Figure 6 displays the RMSF values for the individual amino acids during 50 ns.
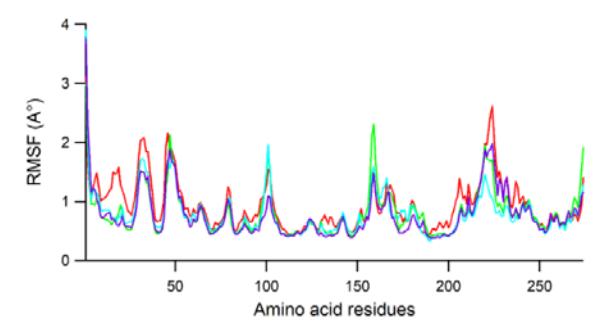
Figure 6 Plot of RMSF for Lig2, Lig10, Lig17 and 1NX, colored red, green, blue, and purple, respectively.
The same pattern of amino acid fluctuation was noted in the whole region of JAK3, which indicates the same binding interaction of the ligands. Overall, the amino acid residues fluctuated under 3 Å, except for those composing the carboxy and amino ends of the protein, which indicates stable dynamics during 50 ns. Dominant peaks were recorded in the residues of around Asp33 (Asp846), HisS46 (His859), Ala101 (Ala919), Asp159 (Asp977), which corresponds to loop regions. The higher peak at Ser224 (Ser1048) is associated with the end of a helix. On the other hand, the amino acid residues involved in the H-bond interactions over all ligands, including Cys91 (Cys909), showed more rigid fluctuation, which indicates protein stability induced by ligand interactions.
Meanwhile, several H-bond interactions of docked conformations were also retained in the MD simulation. For example, H-bonds between Lig2 and JAK3 occurred with occupancies of 45.73%, 26.81%, 16.04%, and 15.72%, with the residues of Arg953 (Arg135), Arg911 (Arg93), Lys830 (Lys17), and Asp967 (Asp149), respectively. Newly formed H-bond interactions with very high occupancies (97.31 and 90.8%) were recorded, namely with Glu871 (Glu58) and Glu85 (Glu903), respectively. On the other hand, Lig10 retained H-bond interactions with Asp912 (Asp94), Lys830 (Lys17), Glu903 (Glu85), Leu905 (Leu87), Gln827 (Gln14) and Lys855 (Lys42), with occupancies of 44.89%, 39.04%, 37.86%, 28.39, 21.54% and 17.36%, respectively. Newly formed Hbond interactions with very high occupancy (71.04 %) were recorded with Glu871 (Glu58) and with moderate occupancy (51.9%) with Arg953 (Arg135). The binding of Lig17 maintained H-bond interaction with Asp967 (Asp149) with 75.72% occupancy, and novel H-bonds were maintained with Arg953
(Arg135) and Asp912 (Asp94) with 88.25% and 54.88% occupancy, respectively. Table S1 shows the H-bond occupancies recorded during the 50-ns simulation.
3.2 Free Binding Energy Calculations
The stable conformation of ligands during the MDs simulation run was then subjected to the MM-PBSA protocol for free binding energy prediction. Prediction of binding energy using the MM-PBSA method is widely considered as more accurate compared to using the docking method. Table 1 shows the calculated binding affinities from the MM-PBSA method. Interestingly, the free binding energies of Lig2 (ΔEPBTOT = −19.35 ± 7.16), Lig10 (ΔEPBTOT = −26.55 ± 8.13), and Lig17 (ΔEPBTOT = −25.34 ± 3.94) were more negative than that of 1NX (−16.90 ± 3.04). The binding energy of Lig17 appeared to be most negative among them. Negative binding free energy originating from favorable electrostatic, van der Waals as well as non-polar energies contributes to the solvation energy.
Ligand ΔEELE (kcal/mol) ΔEVDW (kcal/mol) ΔEPBCAL (kcal/mol) ΔEPBSUR (kcal/mol) ΔEPBTOT (kcal/mol) 1NX −67.65 ± 11.09 −30.53 ± 2.68 85.07 ± 10.94 −3.81 ± 0.20 −16.90 ± 3.04 Lig2 −59.40 ± 13.19 −49.07 ± 5.09 94.70 ± 8.69 −5.58 ± 0.23 −19.35 ± 7.16 Lig10 −91.14 ± 11.90 −57.84 ± 5.10 128.89 ± 15.77 −6.46 ± 0.22 −26.55 ± 8.13 Lig17 −55.88 ± 7.25 −44.37 ± 3.48 79.91 ± 7.20 −5.00 ± 0.13 −25.34 ± 3.94
Tabel 1 MM-PBSA free binding energy for each ligand.
4 Conclusion
Structure-based pharmacophore modelling was performed based on JAK3-1NX interaction. A pharmacophore hypothesis was built and validated. It was then used to identify hit molecules in the Indonesian Herbal Database. Twenty-eight hit molecules were retrieved and then docked into the active site of JAK3 to reveal their binding interactions. The hit molecules maintained their interactions with the JAK3 through H-bond and hydrophobic interactions. The three best hits, i.e. camelliaside B, 3-O-galloylepicatechin-(4beta-6)-epicatechin-3-Ogallate, and mesuaferrone B, were subjected to an MD simulation, which showed good stability during 50 ns. Prediction of binding affinities using the MM-PBSA method showed that the three top hit molecules displayed stronger affinities than 1NX. The contribution of the van der Waals, electrostatic and nonpolar energies to the solvation energy interactions were favorable for most of the three molecules. The present work suggests that these three molecules may serve as potential inhibitors of JAK3.
Acknowledgement
Authors sincerely thanks the Ministry of Research and Technology Republic of Indonesia for its partial funding provided through grant PDUPT, 2020.