
1 Introduction
Kaempferia galanga rhizome is used as a raw material in the traditional medicine industry, especially in Indonesia. It has several functions, for example in the treatment of inflammation as an analgesic, antioxidant, sedative, antimicrobial and even as antineoplastic agent [1]. Essential oil 2.5-4% in the rhizome of K. galanga consists of ethyl cinnamate (25%), ethyl pmethoxycinnamic (30%), p-methoxycinnamic acid, monoterpenes ketone, and 3-carene-5-one. The other compounds are camphene, δ-3-carene, p-methoxy styrene, γ-pinene, β-myrcene, p-cymene, 1,8-cineole, iso-myrcene, camphor, αterpineol, p-cymene- 8-ol, eucarvone and δ-cadinene. Some compounds, such as kaempferol, quercetin, cyanidin and delphinidin, can be found in the leaves of
K. galanga [2]. K. galanga has shown potential in inhibiting cyclooxygenase and TNF-alpha [3], and possibly could also bind to lipoxygenase enzyme because of the active site pockets in cyclooxygenase-2 (COX-2) and 5 lipoxygenase (5-LOX) located near cofactors (Fe2+ in 5-LOX or Hem in COX-2). Nevertheless, the single hydrogen bonds in the interaction of α-amyrins with COX-2 and 5-LOX are formed at the different amino acid residues GLN290 of COX-2 and ASN554 of LOX-5 besides hydrophobic interactions [4]. Antiinflammatory research has been carried out in vitro against isolates of K. galanga such as isopimarane diterpenoids [3].
Lipoxygenases (LOX) is an oxido-reductase enzyme. Single-electron oxidation by iron atom switching between the redox states of Fe2+ and Fe3+ occurs in the LOX catalytic reaction. Arachidonic acid is first oxygenated on the fifth carbon by 5-LOX in conjunction with its activating protein, 5-lipoxygenase-activating protein (FLAP), to give 5-hydroperoxy eicosatetraenoic acid (5-HPETE). 5- LOX, with FLAP, and then catalyzes a second step, the dehydration of 5- HPETE to leukotriene [5].
There are two types of drugs for the treatment of inflammation, non-steroidal anti-inflammatory drugs (NSAIDs) and corticosteroids [6]. Systemic corticosteroids have an integral role in managing inflammatory and immunological conditions. However, these drugs cause serious side effects especially when used at high doses for a long time against conditions such as osteoporosis, adrenal suppression, hyperglycemia, dyslipidemia, cardiovascular disease, Cushing's syndrome, psychiatric disorders, and immunosuppression [7]. Nonsteroidal anti-inflammatory drugs (NSAIDs) are used to reduce pain and inflammation. Based on the level of selectivity for COX inhibition there are two types of NSAIDs, non-selective and COX-2-selective inhibitors (COXIBS) [8]. Long-term use of NSAIDs causes side effects on the gastrointestinal, renal and cardiovascular systems [9].
Zileuton is a drug that can inhibit 5-LOX, an initial enzyme in the leukotriene pathway. Zileuton is marketed for asthma treatment. This drug has limited clinical use because of its hepatotoxicity [10]. Therefore, new drugs are needed that have greater anti-inflammatory activity and fewer side effects than existing anti-inflammatory drugs. Zileuton could be used as a reference in 5-LOX inhibitors studies.
Virtual screening and drug design optimization have been conducted by molecular docking, a method in computer-aided drug design (CADD) [11]. The physical properties of the structure and how the biological macromolecules function can be discovered through molecular dynamics simulation [12], producing root mean square deviation (RMSD) graphs, root mean square fluctuations (RMSF), and potential energy [13][14]. Several docking studies have been conducted to obtain novel 5-LOX inhibitors, for example docking studies of thiazolyl derivatives [15], diospyrin [16] and coumaperine derivatives [17]. This study conducted molecular dynamic simulations of K. galanga compounds as LOX-inhibitor.
2 Materials and Methods
The docking studies in the present study were conducted using the software PLANTS (Protein-Ligand ANT System [18]. The compounds in K. galanga and zileuton in a 3D structure were docked to 5-LOX as receptor. The 3D structure of 5-LOX was downloaded from the Protein Data Bank (PDB) with a resolution of 2.07 Å, PDB code 3V98 [19]. The 5-LOX enzyme was separated from Fe2+ ion previously using YASARA (Yet Another Scientific Artificial Reality Application). First, method validation was carried out by redocking Fe2+ to the receptor with the requirement that the RMSD value was not more than 2 Å [20]. Marvin Sketch transformed the 3D structure of ligands that are available in PubChem into 2D form. Scanning of the process of protonation was carried out by adjusting the pH to 7.4. The YASARA application was used to find the ligand conformation with the lowest energy before proceeding to molecular docking [18]. The Chemical Piecewise Linear Potential (ChemPLP) scores were obtained from the docking result. A more negative value of the ChemPLP score indicates higher affinity of the compound's bond with the receptor. Visualization of the molecular docking results was performed using Discovery Studio. The compound selected to proceed to the molecular dynamic simulation stage was the compound with the most negative ChemPLP score.
Molecular dynamic simulations were carried out using GROMACS (Groningen Machine for Chemical Simulations) on the compound-receptor complex that was selected in the previous stage. The coordinates of this complex were x = 9.08400 Å, y = 12.12800 Å and z = 11.20300 Å. In the molecular dynamic simulation, the LINCS (Linear Constraint Solver) algorithm was used in the AMBER99SB-ILDN ((Assisted Model Building with Energy Refinement) 99SB side-chain dihedral) force field. Molecular dynamics simulation was conducted for 20 ns with temperature at 310 K. The analytical parameters of the simulation were RMSD (root mean square deviation), RMSF (root mean square fluctuations), and potential energy. The molecular dynamics simulation result was visualized using VMD [21].
Figure 1 5-lipoxygenase enzyme with Fe2+ ion from PDB.
3 Results and Discussion
3.1 Data Collection
In this study, 21 compounds of K. galanga flavonoids that have antiinflammatory potential were studied for their activity against the lipoxygenase enzyme. The receptor was the 5-lipoxygenase enzyme with Fe2+ ion (Figure 1) [19].
The drug used as reference ligand was zileuton, which is a 5-lipoxygenase (5- LOX) inhibitor. It is used in asthma treatment and can be used as a selective tool for evaluation of 5-LOX and leukotriene function [22][23].
Zileuton has been studied for various diseases, such as myocardial infarction, non-alcoholic fatty liver disease, and severe respiratory disease [24]-[28]. In PubChem, the 3D structure of the compounds in K. galanga and zileuton were obtained (Table 1) [24].
| No | Compound | PubChem CID | No | Compound | PubChem CID |
|---|---|---|---|---|---|
| 1 | Zileuton | 60490 | 12 | 1,8-cineole | 2758 |
| 2 | Ethyl cinnamate | 637758 | 13 | Iso-myrcene | 519324 |
| 3 | Ethyl p-methoxycinnamic | 5281783 | 14 | Camphor | 2537 |
| 4 | p-methoxycinnamic acid | 699414 | 15 | α-terpineol | 17100 |
| 5 | 3-carene-5-one | 10057675 | 16 | p-cymene-8-ol | 7463 |
| 6 | Camphene | 6616 | 17 | Eucarvone | 136330 |
| 7 | δ-3-carene | 442461 | 18 | δ-cadinene | 12306054 |
| 8 | p-methoxy styrene | 12507 | 19 | Kaempferol | 5280863 |
| 9 | γ-pinene | 6431006 | 20 | Quercetin | 5280343 |
| 10 | β-myrcene | 31253 | 21 | Cyanidin | 128861 |
| 11 | p-cymene | 7463 | 22 | Delphinidin | 128853 |
Table 1 PubChem CID Data of Zileuton and Compounds Contained in K. galanga L.
3.2 Docking Study
Docking studies are part of computational chemistry and can be carried out to understand the interactions that occur between ligands and their receptors, which can predict the bonding association [29]. Docking studies can recognize molecules by finding the active sites on the ligand and the receptor that bind to each other, predicting the binding affinity [30]. Molecular docking in this study was carried out using PLANTS for compounds contained in K. galanga and zileuton as reference ligand. PLANTS applies the flexible docking method, i.e. the receptor and the ligand are in a flexible state in the docking process [29]- [31].
Before beginning the docking study, method validation and redocking were carried out using PLANTS with Fe2+ ion in order to find out the reliability of the docking protocol. The coordinates of the receptor binding sites were obtained by redocking. These coordinates were used as the default receptor binding sites in the virtual screening for 21 compounds of K. galanga and zileuton [32]. The result of redocking 5-LOX with Fe2+ ion produced an RMSD value of 0 Å. The docking method with PLANTS software for 5-LOX as receptor was applicable
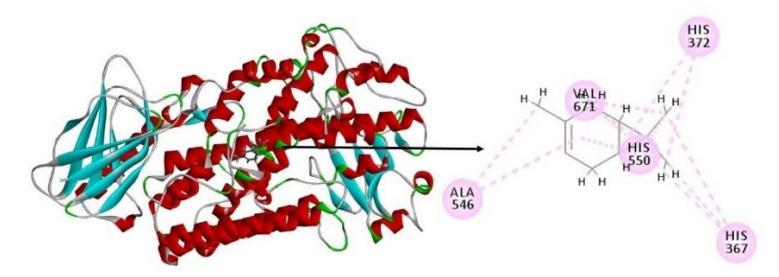
Figure 2 Docking result visualization of δ-3-carene with 5-lipoxygenase complex. δ-3-carene forms 12 hydrophobic bonds with 5-lipoxygenase on the amino acid residues valine 671, alanine 546, histidine 367, histidine 372 and histidine 550.
in this research because the RMSD value of the molecular docking method validation <2 Å [13].
The results are displayed as chemPLP score, which is an empirical fitness function that is optimized for predicting purposes. Among all 21 compounds, the highest affinity was shown by δ-3-carene with the lowest chemPLP score, 20.0002 kcal/mol (Figure 2, Table 2), i.e. higher affinity compared to zileuton. There are 12 hydrophobic bonds between δ-3-carene and 5-lipoxygenase. δ-3 carene and zileuton bind to the same amino acid residue valine 671 in 5-LOX. The interaction types are hydrophobic.
Table 2 Results of Docking Compounds from K. galanga L. and Zileuton to 5 lipoxygenase Enzyme
| No | Compound | ChemPLP Score (kcal/mol) | Bond Type | Binding Site |
|---|---|---|---|---|
| Electrostatic | :UNK*:N2 - :ILE673:O | |||
| Hydrogen Bond | :UNK:H2 - :SER670:O | |||
| 1 | Zileuton | -18.2282 | Hydrophobic | :UNK:C6 - :VAL553 |
| Hydrophobic | :HIS367 - :UNK:C6 | |||
| Hydrophobic | :UNK - :VAL374 | |||
| Hydrophobic | :UNK - :VAL671 | |||
| Hydrogen Bond | :UNK:H5 - :PHE151:O | |||
| 2 | Ethyl cinnamate | -19.9983 | Hydrophobic | :UNK:C5 - :ARG518 |
| Hydrogen Bond | :ARG411:HH12 - :UNK:O3 | |||
| Hydrogen Bond | :UNK:H7 - :THR403:O | |||
| Ethyl p-methoxycinnamic | -19.9923 | Hydrogen Bond | :UNK:H8 - :HIS372:O | |
| Hydrophobic | :UNK:C12 - :ILE415 | |||
| Hydrophobic | :TRP147 - :UNK:C12 | |||
| 3 | Hydrophobic | :PHE151 - :UNK:C12 | ||
| Hydrophobic | :HIS372 - :UNK:C9 | |||
| Hydrophobic | :UNK - :LEU368 | |||
| Hydrophobic | :UNK - :LEU373 | |||
| Hydrophobic | :UNK - :ILE415 | |||
| Hydrogen Bond | :ARG370:H - :UNK:O1 | |||
| 4 | p-methoxycinnamic acid | -19.9995 | Hydrogen Bond | :UNK:H7 - :ILE365:O |
| Hydrophobic | :UNK:C9 - :ARG370 | |||
| Hydrophobic | :UNK:C9 - :VAL553 | |||
| Hydrophobic | :UNK - :LEU443 | |||
| 3-carene-5-one | -20.0001 | Hydrophobic | :UNK - :LEU448 | |
| 5 | Hydrophobic | :UNK:C5 - :LEU369 | ||
| Hydrophobic | :UNK:C5 - :ARG370 | |||
| Hydrophobic | :UNK:C5 - :VAL374 | |||
| Compound | ChemPLP | ||||
|---|---|---|---|---|---|
| No | Score (kcal/mol) | Bond Type | Binding Site | ||
| Hydrophobic | :UNK:C6 - :VAL374 | ||||
| Hydrophobic | :UNK:C6 - :LEU448 | ||||
| Hydrophobic | :UNK:C6 - :MET517 | ||||
| Hydrophobic | :PHE525 - :UNK:C6 | ||||
| Hydrophobic | :PRO152 - :UNK | ||||
| Hydrophobic | :MET517 - :UNK | ||||
| Hydrophobic | :ARG518 - :UNK | ||||
| Hydrophobic | :ARG518 - :UNK | ||||
| Hydrophobic | :UNK - :PRO152 | ||||
| Hydrophobic | :UNK - :MET440 | ||||
| Hydrophobic | :UNK - :MET517 | ||||
| 6 | Camphene | -20.0000 | Hydrophobic | :UNK:C8 - :MET440 | |
| Hydrophobic | :UNK:C8 - :MET517 | ||||
| Hydrophobic | :UNK:C9 - :PRO152 | ||||
| Hydrophobic | :UNK:C9 - :MET440 | ||||
| Hydrophobic | :UNK:C10 - :PRO152 | ||||
| Hydrophobic | :UNK:C10 - :LEU443 | ||||
| Hydrophobic | :UNK:C10 - :MET517 | ||||
| Hydrophobic | :UNK - :VAL671 | ||||
| Hydrophobic | :UNK:C6 - :VAL671 | ||||
| Hydrophobic | :UNK:C7 - :VAL671 | ||||
| Hydrophobic | :UNK:C10 - :ALA546 | ||||
| Hydrophobic | :ALA546 - :UNK | ||||
| Hydrophobic | :HIS367 - :UNK | ||||
| 7 | δ-3-carene | -20.0002 | Hydrophobic | :HIS367 - :UNK:C6 | |
| Hydrophobic | :HIS367 - :UNK:C7 | ||||
| Hydrophobic | :HIS372 - :UNK | ||||
| Hydrophobic | :HIS372 - :UNK:C7 | ||||
| Hydrophobic | :HIS550 - :UNK | ||||
| Hydrophobic | :HIS550 - :UNK:C6 | ||||
| Hydrogen Bond | :HIS367:HD1 - :UNK:O1 | ||||
| Hydrophobic | :UNK:C8 - :LEU414 | ||||
| Hydrophobic | :UNK:C8 - :LEU607 | ||||
| 8 | p-methoxy styrene | -19.9990 | Hydrophobic | :HIS367 - :UNK:C8 | |
| Hydrophobic | :HIS432 - :UNK:C9 | ||||
| Hydrophobic | :UNK - :LEU368 | ||||
| Hydrophobic | :UNK - :LEU414 | ||||
| Hydrophobic | :PRO152 - :UNK | ||||
| Hydrophobic | :ILE365 - :UNK | ||||
| Hydrophobic | :VAL436 - :UNK | ||||
| Hydrophobic | :VAL436 - :UNK | ||||
| Hydrophobic | :ALA439 - :UNK | ||||
| Hydrophobic | :ALA439 - :UNK | ||||
| Hydrophobic | :ALA439 - :UNK:C10 | ||||
| Hydrophobic | :MET440 - :UNK | ||||
| 9 | γ-pinene | -20.0000 | Hydrophobic | :UNK - :MET440 | |
| Hydrophobic | :UNK - :LEU443 | ||||
| Hydrophobic | :UNK:C7 - :ILE365 | ||||
| Hydrophobic | :UNK:C7 - :VAL436 | ||||
| Hydrophobic | :UNK:C8 - :LEU373 | ||||
| Hydrophobic | :UNK:C8 - :VAL436 | ||||
| Hydrophobic | :UNK:C10 - :LEU369 | ||||
| Hydrophobic | :UNK:C10 - :MET440 | ||||
| Hydrophobic | :UNK:C10 - :LEU443 | ||||
| Hydrophobic | :UNK - :LEU373 | ||||
| 10 | β-myrcene | -18.7074 | Hydrophobic Hydrophobic | :UNK:C6 - :MET145 :UNK:C9 - :LEU373 | |
| No | Compound | ChemPLP Score | Bond Type | Binding Site |
|---|---|---|---|---|
| (kcal/mol) | ||||
| Hydrophobic | :TRP147 - :UNK:C6 | |||
| Hydrophobic | :TRP147 - :UNK:C6 | |||
| Hydrophobic | :UNK:C1 - :LEU443 | |||
| 11 | p-cymene | -19.0029 | Hydrophobic | :UNK:C10 - :VAL374 |
| Hydrophobic | :UNK:C10 - :VAL377 | |||
| Hydrophobic | :UNK - :VAL377 | |||
| Hydrogen Bond | :ARG520:HE - :UNK:O1 | |||
| Hydrogen Bond | :ARG520:HH21 - :UNK:O1 | |||
| Hydrophobic | :LEU369 - :UNK | |||
| Hydrophobic | :LEU443 - :UNK | |||
| Hydrophobic | :MET517 - :UNK | |||
| 12 | 1,8-cineole | -20.0000 | Hydrophobic | :UNK - :PRO152 |
| Hydrophobic | :UNK - :LEU369 | |||
| Hydrophobic | :UNK - :LEU443 | |||
| -18.6944 | Hydrophobic | :UNK:C9 - :LEU443 | ||
| Hydrophobic | :UNK:C10 - :MET440 | |||
| Iso-myrcene | Hydrophobic | :UNK:C10 - :LEU443 | ||
| Hydrophobic | :UNK:C10 - :MET517 | |||
| Hydrophobic | :UNK:C9 - :LEU369 | |||
| Hydrophobic | :UNK:C9 - :ARG370 | |||
| Hydrophobic | :UNK:C10 - :VAL553 | |||
| 13 | Hydrophobic | :HIS367 - :UNK | ||
| Hydrophobic | :HIS367 - :UNK:C6 | |||
| Hydrophobic | :HIS367 - :UNK:C10 | |||
| Hydrophobic | :HIS372 - :UNK:C6 | |||
| Hydrophobic | :VAL436 - :UNK | |||
| Hydrophobic | :UNK:C8 - :VAL433 | |||
| 14 | Camphor | -20.0000 | Hydrophobic | :UNK:C8 - :VAL436 |
| Hydrophobic | :PHE151 - :UNK:C8 | |||
| Hydrophobic | :PHE151 - :UNK:C9 | |||
| Hydrophobic | :LEU368 - :UNK | |||
| α-terpineol | Hydrophobic | :VAL433 - :UNK | ||
| Hydrophobic | :UNK:C8 - :ILE365 | |||
| Hydrophobic | :UNK:C8 - :LEU368 | |||
| Hydrophobic | :UNK:C8 - :LEU369 | |||
| 15 | -19.9516 | Hydrophobic | :UNK:C9 - :MET435 | |
| Hydrophobic | :UNK:C9 - :VAL436 | |||
| Hydrophobic | :UNK:C10 - :VAL433 | |||
| Hydrophobic | :TRP147 - :UNK:C10 | |||
| Hydrophobic | :HIS432 - :UNK | |||
| Hydrophobic | :HIS432 - :UNK:C9 | |||
| p-cymene-8-ol | -18.7071 | Hydrophobic | :UNK:C3 - :LEU368 | |
| Hydrophobic | :UNK:C4 - :LEU607 | |||
| Hydrophobic | :UNK:C10 - :ILE406 | |||
| Hydrophobic | :UNK:C10 - :LYS409 | |||
| 16 | Hydrophobic | :HIS367 - :UNK:C3 | ||
| Hydrophobic | :HIS367 - :UNK:C4 | |||
| Hydrophobic | :HIS372 - :UNK:C3 | |||
| Hydrophobic | :HIS372 - :UNK:C4 | |||
| Hydrophobic | :UNK - :ALA410 | |||
| Eucarvone | -20.0001 | Hydrophobic | :PRO152 - :UNK | |
| Hydrophobic | :VAL374 - :UNK | |||
| Hydrophobic | :UNK:C3 - :VAL512 | |||
| 17 | Hydrophobic | :UNK:C3 - :MET517 | ||
| Hydrophobic | :UNK:C4 - :MET517 | |||
| Hydrophobic | :UNK:C10 - :VAL374 | |||
| Hydrophobic | :UNK:C10 - :LEU448 :UNK:C10 - :MET517 | |||
| Hydrophobic |
| No | Compound | ChemPLP Score (kcal/mol) | Bond Type | Binding Site |
|---|---|---|---|---|
| Hydrophobic | :TRP144 - :UNK:C3 | |||
| Hydrophobic | :PHE378 - :UNK:C10 | |||
| Hydrophobic | :PHE525 - :UNK:C10 | |||
| Hydrophobic | :LEU368 - :UNK | |||
| Hydrophobic | :UNK:C5 - :LEU373 | |||
| Hydrophobic | :UNK:C5 - :ARG411 | |||
| δ-cadinene | Hydrophobic | :UNK:C14 - :LEU373 | ||
| 18 | -19.8634 | Hydrophobic | :UNK:C15 - :LEU373 | |
| Hydrophobic | :TRP147 - :UNK:C5 | |||
| Hydrophobic | :TRP147 - :UNK:C5 | |||
| Hydrophobic | :HIS372 - :UNK:C15 | |||
| Hydrogen Bond | :ARG411:HH11 - :UNK:O1 | |||
| Hydrogen Bond | :ARG411:HH12 - :UNK:O1 | |||
| 19 | Kaempferol | -17.7609 | Hydrogen Bond | :UNK:O2 - :LEU373:O |
| Electrostatic | :ARG411:NH2 - :UNK | |||
| Electrostatic | :GLU376:OE2 - :UNK | |||
| Quercetin | -16.9874 | Hydrogen Bond | :ILE155:HN - :UNK:O6 | |
| Hydrophobic | :TRP147 - :UNK | |||
| 20 | Hydrophobic | :TRP147 - :UNK | ||
| Hydrophobic | :UNK - :LEU373 | |||
| Hydrogen Bond | :UNK:H7 - :HIS432:O | |||
| Cyanidin | Hydrogen Bond | :VAL436:HA - :UNK:O3 | ||
| Hydrogen Bond | :LEU369:HN - :UNK | |||
| 21 | -13.3429 | Hydrophobic | :UNK - :ILE365 | |
| Hydrophobic | :UNK - :LEU369 | |||
| Hydrophobic | :UNK - :VAL436 | |||
| Delphinidin | Hydrogen Bond | :UNK:H9 - :THR364:O | ||
| Hydrophobic | :LEU373:HD12 - :UNK | |||
| 22 | -13.3238 | Hydrophobic | :UNK - :LEU369 | |
| Hydrophobic | :UNK - :MET440 | |||
| Hydrophobic | :UNK - :MET440 |
*UNK = Ligand
3.3 Molecular Dynamics Simulation
The stability of the δ-3-carene and 5-lipoxygenase complex was assessed by molecular dynamics simulation (Figure 3). The backbone of both molecules could bend and move easily in the molecular dynamics process [33][34]. The simulation was performed for 20 ns at 310 K.
The relationship between the RMSD value and the time length for the molecular dynamics ascertain the stability of the interactions between the ligand and
Figure 3 The molecular dynamics visualization for the δ-3 carene with 5-lipoxygenase complex.
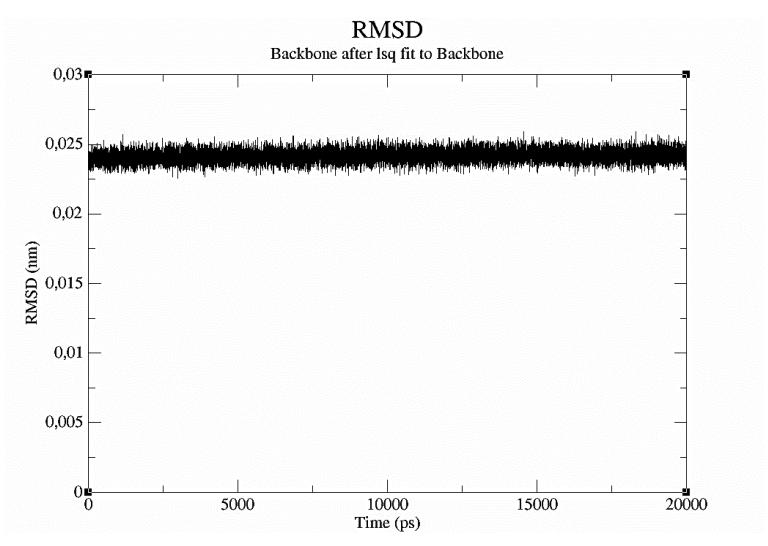
Figure 4 Graph of RMSD changes with time during the molecular dynamic simulation of δ-3-carene with 5-lipoxygenase complex.
receptor molecules. The molecular dynamics result was an RMSD value for the interaction between δ-3-carene and 5-lipoxygenase of less than 0.03nm (0.3Å) (Figure 4). This represents a stable complex structure [35]. The RMSF value indicates the atomic movement of the protein during the molecular dynamic simulation. In the RMSF graph, the atoms of the receptor that fluctuate more were the 1st atom (N of serine; RMSF 0.0388 nm), the 263rd atom (C of glycine; RMSF 0.0310nm), the 3045th atom (C of glycine; RMSF 0.0307nm), the 3430th atom (C of glycine; RMSF 0.0316nm), and the 4431st atom (C of glycine; RMSF 0.0309nm) (Figure 5). Potential energy illustrates the energy stability during a molecular dynamic simulation [14]. The graph of the potential energy complex of δ-3-carene with lipoxygenase showed energy stability during the simulation with a mean of -1.667392 x 106 kcal/mol (Figure 6).
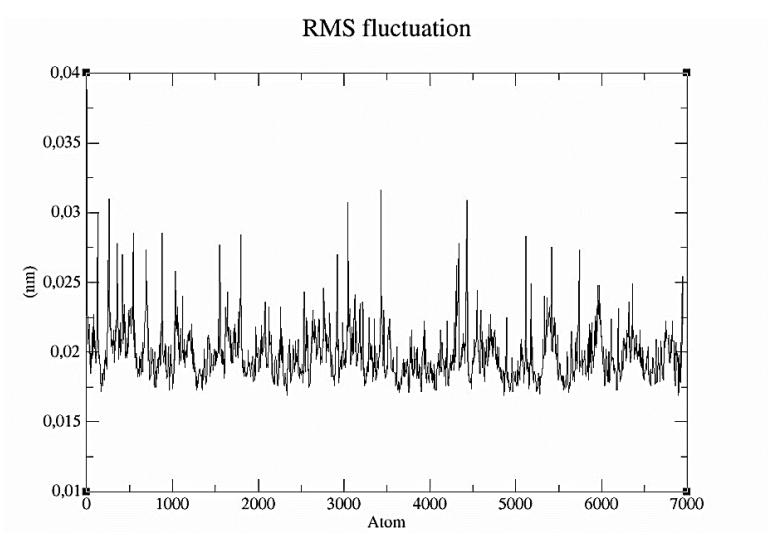
Figure 5 Graph of RMSF in the molecular dynamic simulations of δ-3-carene with 5-lipoxygenase complex.
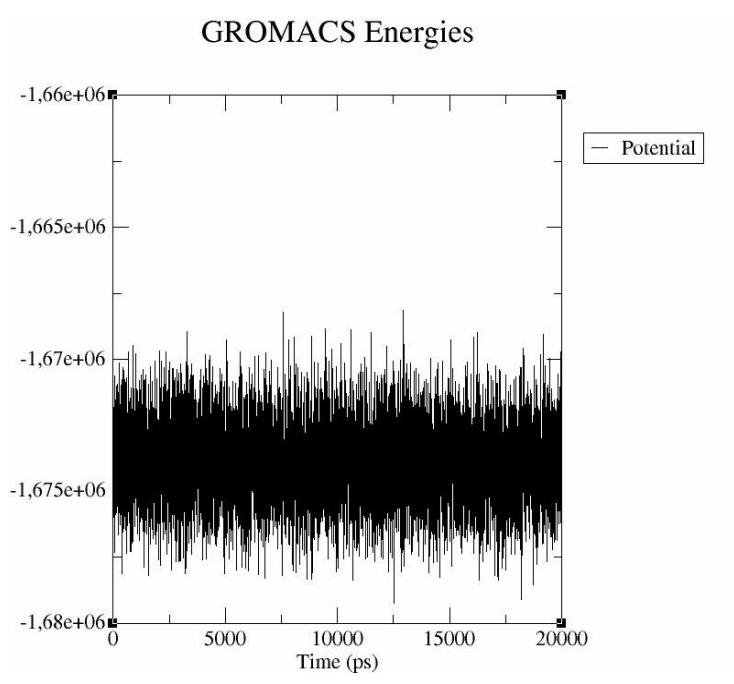
Figure 6 Graph of potential energy in the molecular dynamic simulations of δ-3-carene with 5-lipoxygenase complex.
4 Conclusion
The compound in K. galanga L. that has the greatest affinity in inhibiting the enzyme 5-lipoxygenase (5-LOX) is δ-3-carene with the most negative ChemPLP value, -20.0002 kcal/mol. The binding affinity of δ-3-carene to the 5- LOX receptor is higher than with zileuton as a reference ligand. The complex had good stability during the molecular dynamic simulation with an RMSD and RMSF value smaller than 3Å and -1.67392 x 106 kcal/mol for the mean of potential energy with a simulation time of 20ns.
Acknowledgements
The authors acknowledge the support received from the Ministry of Research, Technology and Higher Education of the Republic of Indonesia.