
1 Introduction
One of the main challenges in the commercialization of hydrogen fuel cell technology is the high usage of the scarce metal platinum (Pt) as an oxygen reduction reaction (ORR) catalyst at the cathode of the fuel cell. The intensive demand for Pt hinders the widespread adoption of fuel cell technology. Hence, substituting the Pt catalyst with a non-precious metal catalyst (NPMC) is believed to be a viable solution for solving this commercialization problem [1]-[3]. Metalorganic frameworks (MOFs) are one class of NPMCs that may answer the ORR
<sup>2</sup> Research Center for Nanosciences and Nanotechnology, Institut Teknologi Bandung, Jl. Ganesha 10, Bandung 40132, Indonesia
<sup>3</sup> Engineering Physics Department, Institut Teknologi Sumatera Jl. Terusan Ryacudu, Way Huwi, Lampung 35365, Indonesia *E-mail: nuruddin@itb.ac.id
challenges. Metal-organic frameworks (MOFs), also referred to as coordination polymers, are crystalline nanoporous materials that have a high surface area and are composed of metal centers and organic ligands [4],[5]. MOFs are typically formed when the organic ligands donate a pair of electrons to the metal cations, filling the metal cation orbitals. This hybrid framework structure allows researchers to customize both the pore size and the chemical environment, enabling the design of different types of targeted performance [6]. Thus, MOFs can be adjusted to have good electrochemical and electrocatalytic activity [7]. In the last decade, a large number of studies on MOFs have been carried out [8] due to their appealing network structures and potential applications in drug delivery [9],[10], luminescent sensing [11], gas storage and separation [12],[13], and catalysis [4],[14],[15]. One of the first MOFs used for ORR catalyst application was the Cu-BTC [16]. The Cu-BTC is a type of MOF that has a square paddlewheel cage structure. This structure has open metal sites (with two Cu atoms at its center) and low structural complexity. While this material can catalyze the ORR through a four-electron reduction mechanism, its ORR activity is still much lower than that of Pt-based catalysts.
Gallis et al. [17] investigated how the molecule of oxygen interacts with TM-BTC MOF (TM = Cu, Mn, Fe, Co). They found that the TM-BTC MOF can interact well with the O2 molecule, and molecular adsorption occurs at the open metal site of the square paddle-wheel cage structure of the MOF. This finding suggests that the TM-BTC MOF is a potential candidate for an ORR catalyst, as the adsorption of the O2 molecule is the first fundamental stage and one of the most crucial steps in ORR. However, the low electrical conductivity of TM-BTC prevents its application as a proper electro-catalyst. Fleker et al. [18] solved this problem by incorporating the MOF inside a conducting matrix of mesoporous activated carbon (Cu-BTC@AC), resulting in high electrical conductivity of the MOF composite. A few years later, Gonen et al. [19] reported on the ORR activity of TM-BTC catalyst incorporated in activated carbon. They unexpectedly found that the Mn-BTC system had the best ORR performance among the other TM-BTC MOFs. This trend is somehow different from the common NPMC system, which generally prefers Fe as the most active metal. Unfortunately, the reason why the TM-BTC system can produce such a trend is still insufficiently understood.
To solve this issue, in this work, we performed a comparative study of the ORR mechanism on the open metal site of the square paddle-wheel cage structure of TM-BTC (TM = Mn, Fe, and Co) MOFs to elucidate the trend of their ORR activities, employing density functional theory (DFT) computations in conjunction with microkinetic simulations. We first studied the interaction of ORR-related adsorbates with the TM-BTCs and the possible ORR mechanisms in these systems. The turn-over frequency (TOF) for ORR was then determined by microkinetic modeling using the energetical information from DFT calculations as the input. By using this method, we could directly compare the ORR activities of the TM-BTC systems.
2 Computational Details
The primitive unit cell of the TM-BTC system is huge and contains hundreds of atoms. In this study, we simplified the calculation by using a small cluster model for the main active site structure of the TM-BTC. Several studies have shown that the open metal site of the square paddle-wheel cage structure of TM-BTC can easily interact with various adsorbates [17],[20]-[22]. Therefore, we assumed that the ORR process in TM-BTC systems will occur on this paddle-wheel site. The simplest cluster model for the square paddle-wheel cage structure that was used in this study is shown in Figure 1. The ORR process was studied on this cluster model. Oversimplification of the TM-BTC active site structure was expected to cause some discrepancies with the full system. However, since all TM-BTC interactions with ORR intermediates occur on the same active site structure, we believe that our simple cluster model would be sufficient to reproduce the main activity trend from the experiment.
Figure 1 (a) Top view and (b) side view of the square paddle-wheel cage active site model of the TM-BTC system.
All DFT calculations were carried out using the Gaussian 09 package [23]. We computed the energy using the triple-zeta 6-311++G(d,p) basis set and the B3LYP exchange-correlation functional. The paddle-wheel cage cluster with transition metal centers is a spin-polarized system but because all of the calculated ⟨ 2 ⟩ values differ by less than 10% from the corresponding ideal values, spin contamination is thought to be sufficiently minor and will have no effect on bond lengths [24]. For the atomic charge calculations, we employed the natural bond orbital (NBO) population scheme. The entire population of valence electrons on each target atom was subtracted to obtain the difference in electron population between that atom or molecule before and after O2 adsorption (Δ2 ).
The adsorption energy of an ORR intermediate on the paddle-wheel cage active site of TM-BTC (\(E_{mol}^{ad}\)) is defined as:
\[E_{mol}^{ad} = E_{tot} - E_{TM-BTC} - E_{mol} \tag{1}\] where \(E_{tot}\) represents the total energy of a combined system, \(E_{TM-BTC}\) represents the total energy of the paddle-wheel cage cluster, and \(E_{mol}\) represents the total energy of an isolated ORR intermediate. The corresponding spin configurations for the \(E_{tot}\), \(E_{TM-BTC}\) and \(E_{mol}\) calculations were put in the associated spin ground states. In this formulation, steady adsorption and negative adsorption energy are synonymous.
Typical ORR mechanism in an alkaline medium consists of the following elementary reactions [25]-[28]:
\[O_2 + * \rightarrow *O_2 \tag{2}\]
\[^*O_2 + H_2O + e^- \rightarrow ^*OOH + OH^-\] (3)
\[^*OOH + e^- \rightarrow ^*O + OH^-\] (4)
\[^*O + H_2O + e^- \rightarrow ^*OH + OH^-\] (5)
\[^{*}OH + e^{-} \rightarrow * + OH^{-} \tag{6}\]
Reaction (2) is a chemical reaction (\(O_2\) adsorption), while reactions (3)-(6) are electrochemical reactions. All of these reactions occur on top of the TM atom of the paddle-wheel cage active site of the TM-BTC. *X and * correspond to an adsorbed X molecule and a free adsorption site. Using the aforementioned reactions, we need to create an ORR free energy diagram to comprehend the trend of ORR activity on TM-BTC systems.
The free energies for the electrochemical reactions were calculated using the computational hydrogen electrode (CHE) approximation [29]. In the CHE approximation, the chemical potential for the \([H^+ + e^-]\) term under standard conditions can be connected to the chemical potential of half of an \(H_2\) molecule by referencing them to the standard hydrogen electrode. Using this assumption, the following relation may be used to compute the change in free energy \(\Delta G(U)\) for reaction \(*A + H+ + e-(U) \rightarrow *AH\) at bias potential U:
\[\Delta G(U) = E(*AH) - E(*A) - \frac{1}{2}E(H_2) + \Delta ZPE - T\Delta S\] (7)
- pHkbT ln 10 + eU, where \(E(^*X)\) and E(X) are the total energies of an adsorbed X species and an isolated X species respectively, as determined by DFT calculations. \(\Delta\)ZPE is the shift in zero-point energies and \(\Delta S\) represents the entropy change. The NIST database was used to determine the zero-point energies and the entropies of the ORR intermediates.
The main parameters we used for assessing the ORR activity of the TM-BTC catalyst were overpotential (\(\eta\)) and onset potential (\(U^{onset}\)) [29]. A catalyst system with better ORR activity will have a lower \(\eta\) or a higher \(U^{onset}\). The \(U^{onset}\) is the lowest electrode potential at which one of the elementary electrochemical reactions has free energy differences that equal zero, \(\Delta G(U^{onset}) = 0\). The ORR free energy diagram at \(U > U^{onset}\) will have at least one elementary electrochemical with an uphill profile (endergonic). The ORR overpotential is related to the onset potential as expressed in the following equation:
\[\eta = U^{eq} - U^{onset} = \Delta G_{max}(U^{eq})/e\] (8)
where \(U^{eq}\) is equivalent to the ORR equilibrium potential and \(\Delta G_{max}(U^{eq})\) is the maximum \(\Delta G\) of elementary reactions at \(U = U^{eq}\). From Equation (8) it is evident that \(U^{onset}\) and \(\eta\) are linearly dependent on one another, hence, they may both be used as measures of ORR activity.
Under the same standard conditions (pH = 0, p = 1 bar, T = 298.15 K, and U = 0V), the Gibbs free energy profiles for ORR elementary reactions in alkaline and acidic are identical [27],[28]. Additionally, the RHE potential also makes up for the pH effect's inclusion in the chemical potential of H+. This results in an equalized computed overpotential for ORR on acidic and alkaline media within the CHE approximation. As a consequence, the ORR activity trend determined by this approximation holds true for both acidic and alkaline media. We also neglected the effect of solvation in our model. Since the adsorption site for the TM-BTC active sites is identical, the TM-BTC active sites will have similar adsorption configurations for ORR intermediates. Therefore, the effect of a solvent on the free energy of adsorption on TM-BTC active sites will be similar and will not affect the trend of molecular adsorption energy on these sites [30].
To determine the turnover frequency (TOF) of the ORR process, the computed ORR free energies were fed into a microkinetic simulation. The TOF value was used to evaluate the kinetic of the ORR process on TM-BTC systems. The details of the microkinetic simulation can be found in our recent publications [30]-[38].
3 Results and Discussions
First, we discuss the adsorption of the O<sub>2</sub> molecule on the paddle-wheel active site of TM-BTC systems. This step is very important, as it will determine whether the ORR can proceed or not. The energetical and geometrical parameters of the most stable O2 adsorption configurations on the active sites of TM-BTC are presented in Table 1 and Figure 2.
Table 1 Energetical and geometrical parameters of the most stable O2 adsorption configurations on the active sites of TM-BTC systems.
| System | O-O | TM-O2 | TM-O-O | 𝚫𝑸𝑶𝟐 | 𝒂𝒅 𝑬𝑶𝟐 |
|---|---|---|---|---|---|
| (Å) | (Å) | o ( ) | (e) | (eV) | |
| Mn-BTC-O2 | 1.217 | 2.160 | 125.80 | -0.16 | -0.22 |
| Fe-BTC-O2 | 1.244 | 2.150 | 118.26 | -0.23 | -0.22 |
| Cu-BTC-O2 | 1.205 | 2.673 | 126.87 | 0.05 | -0.13 |
| Isolated O2 | 1.206 | - | - | - | - |
Figure 2 Adsorption configurations of the O2 molecule on the square paddlewheel active site of (a) Mn-BTC, (b) Fe-BTC, and (c) Cu-BTC.
The O2 molecule is adsorbed on all the TM-BTC systems with an end-on configuration where only one O atom of the O2 molecule is bonded to the TM atom of the paddle-wheel cage active site. This configuration is a typical O2 adsorption configuration on TM-Nx active sites [39]-[46]. In general, we find that the Mn-BTC and Fe-BTC active sites have stronger interaction with the O2 molecule as compared to that on the Cu-BTC active site. This condition is indicated by the values of O2 adsorption energy, extra charges in O2 (Δ2 ), and O-O bond elongation. The length of the O-O bond and the amount of charge transferred to the adsorbed O2 molecule increase with the strength of O2 adsorption [40],[42],[46].
In general, the interaction between the anti-bonding orbital (2π*) of the O2 molecule and the cluster's frontier molecular orbitals (fMOs), which have strong dz2-like orbital characteristics on their TM site, facilitates the development of O2 end-on adsorption on the TM atom [42]. Following this adsorption, some charges (Δ2 in Table 1) are transferred from the TM-BTC paddle-wheel cage cluster to the adsorbed O2 molecule. In the Mn and Fe-BTC cases, a significant number of extra charges reside in the adsorbed O2 molecule (indicated by the negative value of Δ2 ). These extra charges occupy the half-filled 2π* orbitals of the O2 molecule and increase the repulsion between the O atoms in the adsorbed O2 molecule [40],[42],[46]. This repulsion leads to O-O bond elongation of the adsorbed O2 molecule on Mn and Fe-BTC systems. In the Cu-BTC case, no extra charges are transferred from the Cu atom to the adsorbed O2 molecule. Because of this, there is only a very weak Cu-O2 interaction, as seen by the low O2 adsorption energy and the absence of O-O bond elongation relative to the isolated O2 molecule. This trend is quite similar to the case of H2 adsorption on the paddlewheel active site of TM-BTC [20]. This trend occurs because the dz2-like orbital lies in a deeper energy level than that in the other systems, resulting in the full occupancy of anti-bonding states between Cu and the molecule [20]. The full occupancy of this anti-bonding orbital weakens the Cu-molecule interaction as compared to the partial occupancy in the other systems. Since the Cu-BTC system has weaker O2 adsorption energy than the Mn-BTC and Fe-BTC systems, we expect that the Cu-BTC will also have weaker interaction with other ORR intermediates.
After confirming that the O2 molecule can be adsorbed on the TM atom site of the paddle-wheel active site of TM-BTC systems, we are now certain that subsequent elementary reactions of the ORR may continue on these systems. Next, we calculate the adsorption of ORR intermediates involved in elementary reactions (2)-(6) on the paddle-wheel active site of the TM-BTC system and construct their ORR-free energy diagrams. Figure 3 displays the adsorption geometries of the ORR intermediates. The ORR free energy profiles at zero electrode potential and equilibrium potential are presented in Figures 4a and 4b, respectively. From the profile at zero potential, it can be seen that the majority of the elementary reactions on the paddle-wheel active site of the Mn-BTC and Fe-BTC systems have downhill profiles (exergonic), while the reactions on the Cu-BTC system have a noticeable uphill profile (endergonic) for the O2 reduction to OOH step (Reaction 3). The endergonic profile for this elementary step indicates that the OOH adsorption on the active site of the Cu-BTC system is much weaker than for the other two systems. This trend is expected since we already know that the Cu-BTC-O2 interaction is weaker than in the other systems. Since such an uphill profile for the O2 reduction to OOH already occurs at zero potential bias, this elementary step will hamper the ORR process from the beginning. Therefore, this elementary step will act as the main thermodynamics determining step (TDS) for ORR in the Cu-BTC system.
In contrast to the Cu-BTC system, the TDS for Mn-BTC and Fe-BTC systems originate from the OH reduction to OH- (Reaction 6) because this elementary step has the smallest Δ in the ORR profile at zero potential bias. A larger applied potential bias will give this reduction step an uphill profile. In general, a very
small value of Δ for the OH reduction to OHindicates that a system has a quite strong OH adsorption energy [44],[45]. This trend also applies to the TM-BTC system because the OH adsorption energy in the Mn-BTC and Fe-BTC systems (-2.68 eV and -2.73 eV) is much stronger than in the Cu-BTC system (-0.04 eV). Again, this result is expected, since the Mn and Fe-BTC systems have stronger interaction with adsorbate than the Cu-BTC system.
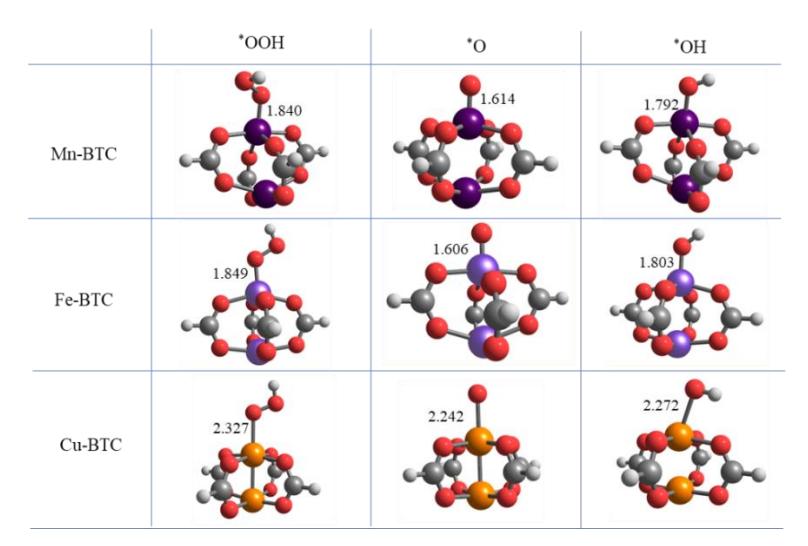
Figure 3 Adsorption configurations of ORR-related intermediates on the square paddle-wheel active site of TM-BTC systems.
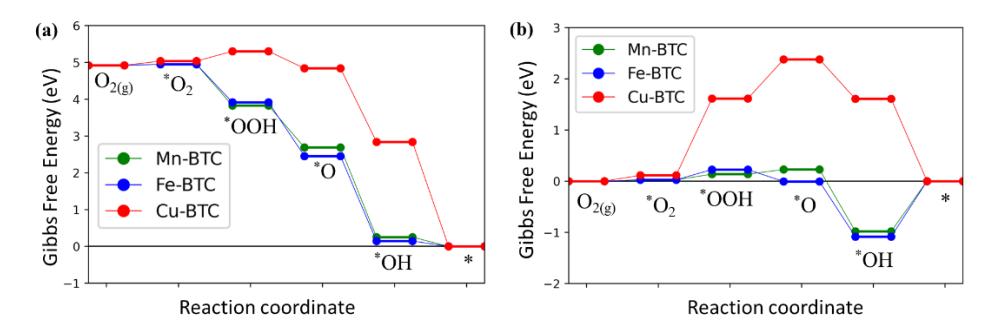
Figure 4 ORR free energy profiles on the square paddle-wheel active site of TM-BTC systems at zero bias and equilibrium potential.
After knowing the TDS candidate for the TM-BTC systems, we can easily determine the value of overpotential (η) for ORR in these systems by calculating the value of Δ for the TDS at equilibrium potentials, as indicated in Figure 4b [29]. The value of onset potential ( onset) can be calculated directly by using Equation (8). The values of onset potentials and overpotentials for TM-BTC
systems are presented in Table 2. The relative value of the calculated onset potential and overpotential from the CHE method can be used to make a rough estimation of the ORR activity of a catalyst. A smaller value of overpotential (or larger onset potential) for the ORR through an associative reduction mechanism corresponds to higher ORR activity [29]. From the trend in Table 2, we can see that the approximate activity trend for the ORR on TM-BTC is: Mn-BTC > Fe-BTC > Cu-BTC. To make a more proper judgment for the ORR activity trend, we need to perform microkinetic simulations to obtain the value of turnover frequency (TOF) with respect to applied electrode potential U. We used the free energy data from the CHE method as the input for our microkinetic simulation.
Table 2 Adsorption energies of ORR intermediates and the calculated onset potentials and overpotentials from CHE approximation on square paddle-wheel active site TM-BTC systems.
| System | EO2 (eV) | Ead00H (eV) | EadOH (eV) | Onset potential Uonset (V) | Overpotential \(\eta\) (V) |
|---|---|---|---|---|---|
| Mn-BTC | -0.22 | -1.71 | -2.68 | 0.25 | 0.98 |
| Fe-BTC | -0.21 | -1.72 | -2.73 | 0.15 | 1.08 |
| Cu-BTC | -0.13 | -0.24 | -0.04 | -0.27 | 1.50 |
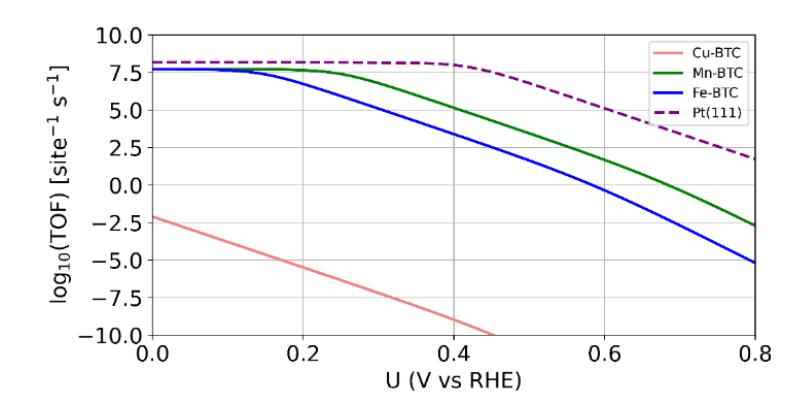
Figure 5 TOF profiles as a function of bias potential for ORR on the square paddle-wheel active site of TM-BTC systems.
Figure 5 shows the TOF profiles for ORR on TM-BTC systems as a function of applied electrode potential U. A larger TOF value corresponds to better ORR activity. For comparative purposes, we also include the TOF profile for the Pt(111) surface as a benchmark. Pt(111) is the most active surface for the ORR process. The TOF for the Pt(111) system was recalculated by using the free energy data from Ref. [29] with \(O_2\) adsorption energy equal to -0.46 eV (range of \(O_2\) adsorption energy on Pt(111): -0.46 to -0.86 eV [47]-[50]). We can see that the TOFs of the Mn-BTC and Fe-BTC systems are superior to that of the Cu-BTC system. This trend is also consistent with the overpotential/onset potential
trend from the CHE approximation. We can also see that the TOF of the Pt(111) system is still better than that of the TM-BTC systems. This trend is in complete agreement with the experimental trend from the work of Gonen et al. [19]: Pt > Mn-BTC > Fe-BTC > Cu-BTC. This result shows that the simple paddle-wheel active site model is appropriate for studying the ORR activity trend of TM-BTC systems.
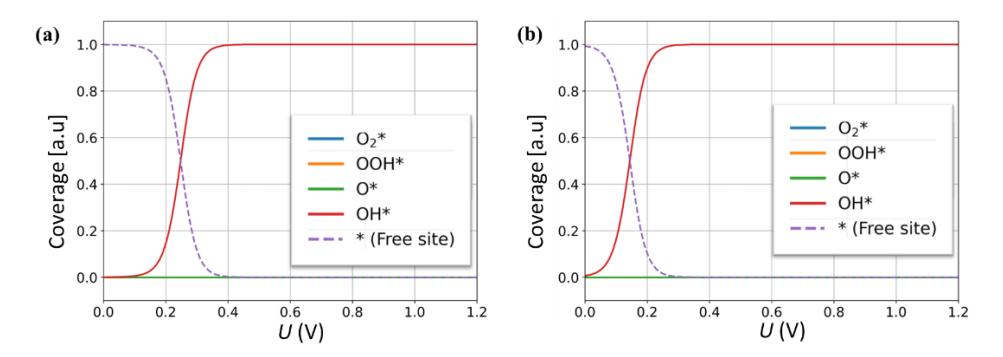
Figure 6 The surface coverage profiles of ORR intermediates as a function of bias potential in (a) Mn-BTC and (b) Fe-BTC systems.
After successfully reproducing the experimental trends for the TM-BTC system, we can now give some comments on why the Mn-BTC system has better ORR activity than the Fe-BTC system as observed in the experiment. This issue can be traced back to the origin of overpotential/onset potential in these two systems. From Figure 5, we can see that the TOF profiles for the Mn-BTC and Fe-BTC systems start to decrease around their CHE onset potentials. This shows that the TOF drop is directly related to the CHE onset potentials and is determined by the ORR thermodynamic determining step (TDS) of each active site configuration. From the CHE approximation, we know that the TDSs in these systems both originate from the OH reduction step (Reaction 7). The Mn-BTC system has a smaller overpotential (or larger onset potential) because the Δ of its TDS (OH reduction step) at the equilibrium potential (Figure 4b) is smaller than in the Fe-BTC system. As mentioned above, this can also be read as the Mn-BTC system having weaker OH adsorption energy than the Fe-BTC system (-2.68 eV versus -2.73 eV, see Table 2). Therefore, the reason why Mn-BTC has better ORR activity, as observed in the experiment, is that it has weaker OH adsorption energy than the Fe-BTC system. This conclusion is reasonable because stronger OH adsorption will make the OH reduction step more sluggish since it requires extra effort to release the OH from the active site. This can also be easily confirmed by inspecting the profiles of surface coverage for the Mn-BTC and Fe-BTC systems, as shown in Figure 6. It is clear that at > onset, the active sites are fully covered by OH intermediates, and the TOF becomes very small in this potential
region due to the sluggish OH reduction step. The Mn-BTC system has a better TOF because the increase of OH coverage occurs at a higher bias potential than in the Fe-BTC system.
In the previous discussion, we have shown that our simple cluster model could successfully reproduce the experimental ORR activity trend for TM-BTC systems. However, there were some expected problems in our result. The absolute values for the overpotentials/onset potentials were still smaller than the experimental values, and the TOF value for the Cu-BTC system was much smaller than for the other TM-BTC systems. The cause of these problems might come from oversimplification of the TM-BTC structure and the exclusion of the active carbon model. The absence of the full MOF structure and active carbon might reduce the ability of the Cu paddle-wheel cage active site to transfer electrons and hence weaken the Cu interaction with ORR-related intermediates. We speculate that the inclusion of a full Cu-BTC structure may stabilize the OOH adsorption on the active site and subsequently improve the value of its overpotential and TOF.
4 Conclusion
We have studied the ORR mechanism on the active site of TM-BTC (TM = Mn, Fe, Cu) MOFs by using a combination of DFT and microkinetic calculations. The ORR process in TM-BTC systems was studied by using a simple small cluster model, which represented the structure of the square paddle-wheel cage active site of the TM-BTC systems. By using this simple model, we successfully reproduced the experimental ORR activity trend from Gonen et al. [19], Pt > Mn-BTC > Fe-BTC > Cu-BTC. The Cu-BTC system has the lowest ORR activity because its active site cannot properly bind OOH intermediates, which results in a quite high ORR overpotential. We also showed that the unusual ORR activity trend for Mn and Fe systems (Mn-BTC > Fe-BTC), as observed from the experiment, originates from the trend of their OH adsorption energies, which also determines their ORR overpotential. The Mn-BTC system has better ORR activity than the Fe-BTC system because it has weaker OH adsorption. A very strong OH adsorption makes the final OH reduction step sluggish, hence hindering the ORR process.
Acknowledgments
This research was fully funded by the PPMI 2021 program at the Institut Teknologi Bandung. The high-performance computing facility at the Research Center for Nanosciences and Nanotechnology at the Institut Teknologi Bandung was used for some of the calculations.