
Pendahuluan
Saat ini, obat golongan diuretik sering disalahgunakan oleh atlet olahraga dalam berbagai event olah raga, terutama pada cabang olahraga dengan parameter berat badan sebagai acuan perlombaan. Beberapa atlet menurunkan berat badan secara cepat dengan mengkonsumsi senyawa-senyawa diuretik dengan tujuan agar dapat masuk ke dalam klasifikasi berat badan yang diinginkan. Disamping bertujuan menurunkan berat badan, beberapa atlet olahraga menggunakan senyawa diuretik untuk menutupi penggunaan doping, dengan mengencerkan sehingga konsentrasi senyawa doping menjadi sangat rendah dan tidak terdeteksi saat pemeriksaan urin.
Komisi Medik IOC (International Olympic Commitee) melarang penggunaan senyawasenyawa diuretik dalam setiap pertandingan olimpiade. Kemudian WADA (World Anti-Doping Agency), yang diprakarsai oleh IOC, dibentuk dengan tujuan untuk memerangi penyalahgunaan obat-obatan dalam olahraga, dan membantu federasi-federasi olahraga untuk melakukan prosedur pengujian, serta menerbitkan daftar yang berisi zat yang dilarang untuk dikonsumsi seorang atlet.
Diuretik adalah zat-zat yang dapat meningkatkan atau memperbanyak pengeluaran air kemih (diuresis) melalui kerja langsung terhadap ginjal. Diuretik pada umunya diklasifiskasikan dalam kelompok loop diuretik (furosemid), derivat tiazid (hidroklorotiazid), serta diuretik hemat kalium (spironolakton).
Beberapa peneliti telah berhasil mengembangkan metode analisis yang dapat digunakan untuk pengujian hidroklorotiazid, furosemid, dan spironolakton dalam urin (Zendelovska, 2006). Namun demikian metode tersebut masih memiliki keterbatasan sehingga peluang pengembangan metode analisis zat ini masih terbuka. Penggunaan suatu metode standar yang telah tervalidasi akan memberikan data analisis yang akurat dan presisi dalam waktu yang relatif singkat.
Dilatarbelakangi oleh beberapa hal di atas, penelitian ini bertujuan untuk mendapatkan suatu metode kromatografi cair kinerja tinggi (KCKT), yang dapat digunakan untuk menganalisis hidroklorotiazid, furosemid, dan spironolakton dalam urin secara simultan.
Percobaan
Bahan
Bahan-bahan yang dipakai dalam penelitian ini meliputi baku pembanding hidroklorotiazid, furosemid, dan spironolakton (Merck), urin manusia, asetonitril pro HPLC (JT Baker), aquabidestilata, dinatrium hidrogen fosfat, etil asetat, amonium klorida dan bahan lain yang biasa dipakai di laboratorium farmasi analisis.
Alat
Alat-alat yang dipakai dalam penelitian ini meliputi Kromatografi Cair Kinerja Tinggi (HPLC Agilent HP 1100), kolom LichroCART C18 (4,0 mm, 250 mm, 10 m), spektrofotometer UV-Vis (Beckman Du 650i), pH meter (Beckman), timbangan (Mettler AG104), mikropipet, vortex, dan alat gelas yang biasa dipakai di laboratorium farmasi analisis.
Prosedur
1. Penentuan Panjang Gelombang Deteksi pada Daerah Ultraviolet
Larutan baku hidroklorotiazid, furosemid, dan spironolakton diencerkan dengan fase gerak sampai didapat konsentrasi masing-masing sebesar 10 ppm, 5 ppm, dan 20 ppm, kemudian diukur spektrum absorpsi masing-masing senyawa pada rentang panjang gelombang 200-400 nm menggunakan spektrofotometer UV-Vis. Spektrum absorpsi hidroklorotiazid, furosemid, dan spironolakton ditampilkan secara tumpang tindih (overlay) sehingga dapat ditentukan panjang gelombang optimum yang dapat digunakan untuk mendeteksi ketiganya.
2. Penyiapan Sampel
Urin yang akan digunakan disentrifuga terlebih dahulu, lalu dipipet sebanyak 2 mL kemudian dibuat basa dengan menambahkan 200 l larutan 10% dapar salmiak pH 9,5. Kemudian diekstraksi dua kali dengan 5 mL etil asetat. Lapisan etil asetat dipisahkan kemudian diuapkan pada kondisi vakum hingga kering. Residu yang dihasilkan direkonstitusi dengan 200 µl campuran asetonitrildapar fosfat pH 3,0 (1:1) dengan bantuan vortex, lalu disaring menggunakan membran filter 0,45 m.
3. Penentuan Kondisi Optimum Kromatografi Cair Kinerja Tinggi
Pada penelitian ini dilakukan terlebih dahulu penentuan kondisi optimum kromatografi cair kinerja tinggi untuk analisis hidroklorotiazid, furosemid, dan spironolakton. Kondisi optimum
ini didapat dengan pemilihan kolom serta pemilihan komposisi dan pengaturan laju campuran asetonitril-dapar fosfat pH 3 sebagai fase gerak. Penentuan kondisi optimum ini berdasarkan uji kesesuaian sistem dengan menguji parameter resolusi (Rs), selektifitas (\(\alpha\)), faktor kapasitas (k), dan angka lempeng teoritis (N).
4. Uji Kesesuaian Sistem
Uji kesesuaian sistem dilakukan dengan melakukan injeksi larutan standar hidroklorotiazid 10 ppm, larutan furosemid 10 ppm, dan spironolakton 30 ppm masing-masing sebanyak 6 kali. Fase gerak digunakan adalah campuran asetonitril-dapar fosfat pH 3,0 secara gradien, dengan laju alir 1,0 mL/menit, kolom \(C_{18}\) (4,0 mm, 250 mm, 10 \(\mu\)m) pada suhu 25°C, dan detektor pada panjang gelombang 229 nm.
Kondisi elusi gradient
| Waktu | Asetonitril | D. fosfat |
|---|---|---|
| (menit) | (%) | (%) |
| 0 | 13 | 87 |
| 8 | 40 | 60 |
| 11 | 60 | 40 |
| 13 | 80 | 20 |
5. Pengujian Kelinieran, Batas Deteksi, dan Batas Kuantisasi Metode
Larutan seri baku dengan konsentrasi masingmasing 2 ppm, 4 ppm, 6 ppm, 8 ppm, 10 ppm dan 12 ppm untuk hidroklorotiazid dan furosemid serta konsentrasi 5 ppm, 10 ppm, 15 ppm, 20 ppm, 25 ppm dan 30 ppm untuk spironolakton disuntikkan ke dalam gerbang suntik HPLC.
Kelinieran metode dinilai dari kurva hubungan antara AUC dengan konsentrasi, kemudian dihitung persamaan garis regresi linier (Y = ax + b), nilai koefisien korelasi (r) dan koefisien variansi fungsi regresinya (\(V_{xo}\)). Batas deteksi dan batas kuantisasi dihitung dengan Metode Miller (Ibrahim, 2004).
6. Penentuan Perolehan Kembali/Akurasi, dan Presisi Metode
Parameter kecermatan ditentukan dengan cara menghitung persen perolehan kembali melalui spiked-placebo recovery method. Sampel urin yang mengandung 0,5 ppm; 5,0 ppm; dan 10,0 ppm untuk hidroklorotiazid dan furosemid serta 1,0 ppm; 10,0 ppm; dan 20,0 ppm untuk spironolakton digunakan pada pengujian kecermatan metode ini. Masing-masing konsentrasi dianalisis seperti pada penyiapan
sampel dengan 3 kali replikasi dan dihitung perolehan kembali absolutnya dari masing-masing senyawa. Presisi didapat dengan menghitung koefisien variasi (% KV) tiap analit.
7. Penentuan Ketegaran Metode
Penentuan ketegaran metode dilakukan menggunakan cara OFAT (one factor at time) dengan membuat variasi perubahan laju alir fase gerak, perubahan pH dapar fosfat dan perubahan panjang gelombang pada metode normal. Hasil perolehan kembali masing-masing senyawa dari tiap perubahan yang dilakukan dianalisis secara statistik dengan menggunakan metode ANOVA.
Tabel OFAT
| Faktor analisis yang dirubah | Perubahan (-) | Perubahan (+) |
|---|---|---|
| Laju alir (mL/menit) | 0,9 | 1,1 |
| pH dapar fosfat | 2,8 | 3,2 |
| Panjang gelombang (nm) | 227 | 231 |
Hasil dan Pembahasan
Pengembangan metode kromatografi cair kinerja tinggi (KCKT) analisis senyawa diuretik doping dimulai dari penentuan panjang gelombang maksimum dari larutan hidroklorotiazid 10 ppm, furosemid 5 ppm, dan spironolakton 30 ppm. Penentuan panjang gelombang ini dilakukan dengan cara membuat spektrum serapan maksimumnya dengan melakukan penyusuran pada rentang panjang gelombang 200-400 nm. Berdasarkan hasil penelusuran serapan ketiga senyawa tersebut, dipilih panjang gelombang optimum 229 nm karena pada panjang ketiga senyawa gelombang tersebut memberikan serapan dengan absorptivitas yang relatif kuat (\(\varepsilon_{hidroklorotiazid} = 1.8 \times 10^4 \text{ M}^{-1}\text{cm}^{-1}\); \(\varepsilon_{\text{furosemid}} = 3.0 \text{ x } 10^4 \text{ M}^{-1} \text{cm}^{-1}; \ \varepsilon_{\text{spironolakton}} = 1.0 \text{ x } 10^4\)\(M^{-1}cm^{-1}\)).
Optimasi kondisi KCKT ditentukan dengan membuat berbagai kondisi gradien fase gerak dan laju alir fase gerak. Kolom digunakan LichroCART\(^{\otimes}\) C<sub>18</sub> (4,0 mm, 250 mm, 10 \(\mu\)m), suhu 25\(^{\circ}\) C, laju alir 1 mL/menit dan deteksi pada panjang gelombang 229 nm. Untuk mendapatkan profil pemisahan yang baik, kondisi KCKT yang dipilih mengunakan campuran fase gerak
asetonitril dan dapar fosfat pH 3,0 mengikuti kondisi seperti pada Tabel 1.
Hasil elusi dengan menggunakan kondisi di atas, dapat memisahkan hidroklorotiazid, furosemid, dan spironolakton dengan baik. Pemisahan yang dilakukan memenuhi syarat yang telah ditetapkan. Hasil uji kesesuaian sistem dapat dilihat pada tabel 2.
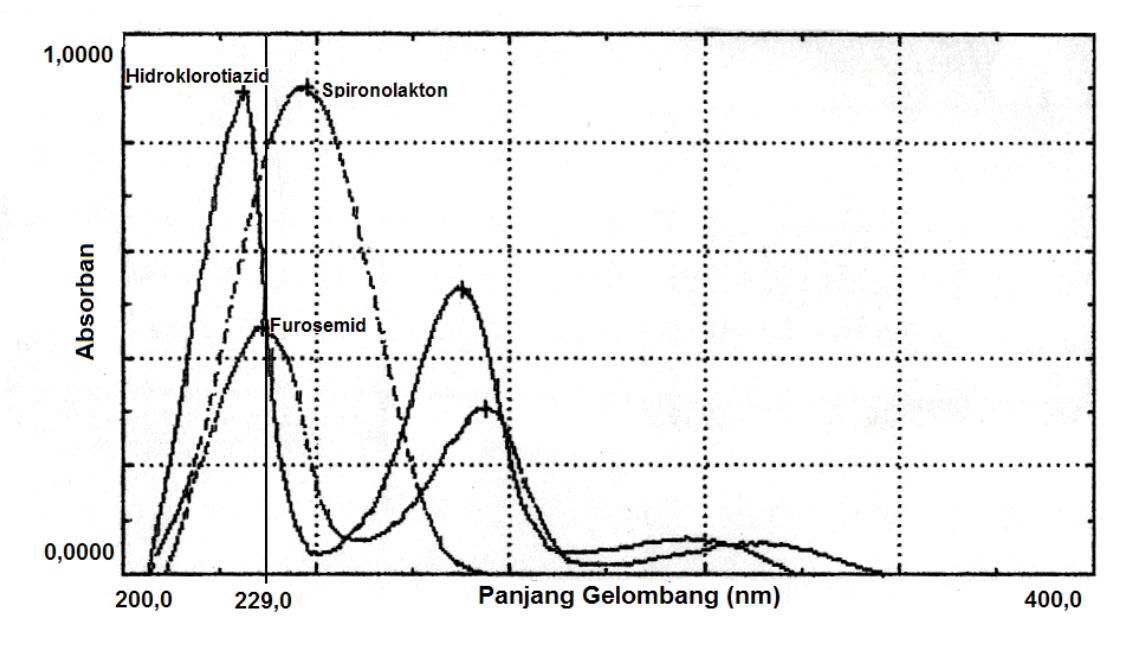
Gambar 1.Spektrum absorpsi UV larutan hidroklorotiazid, furosemid, dan spironolakton
Tabel 2. Uji Kesesuaian Sistem
| Parameter | Nilai | Kriteria penerimaan | |
|---|---|---|---|
| Resolusi (Rs) | |||
| HCT dan Furosemid | 14,6 | Rs 1,5 | |
| HCT dan Spironolakton | 22,9 | ||
| Furosemid dan Spironolakton | 10,3 | ||
| Selektifitas (α) | |||
| HCT dan Furosemid | 2,18 | α 1,5 | |
| HCT dan Spironolakton | 2,84 | ||
| Furosemid dan Spironolakton | 1,31 | ||
| Faktor kapasitas (k') | HCT : 1,94 | 1 k' 10 | |
| FUR : 4,22 | |||
| SPI : 5,50 | |||
| Angka lempeng teoritis (N) | HCT : 21.386/m | N 10.000/m | |
| FUR : 147.392/m | |||
| SPI : 249.914/m | |||
| Keterulangan penyuntikan | |||
| Waktu Retensi | |||
| HCT | KV : 0,14 | KV 2% | |
| Furosemid | KV : 0,05 | ||
| Spironolakton | KV : 0,02 | ||
| AUC | |||
| HCT | KV : 0,55 | KV 2% | |
| Furosemid | KV : 0,44 | ||
| Spironolakton | KV : 0,95 |
Profil kromatogram hidroklorotiazid, furosemid, dan spironolakton baku, blangko urin, dan sampel urin yang dianalisis pada kondisi optimum KCKT dapat dilihat pada gambar 2, gambar 3 dan gambar 4. Pada kondisi KCKT ini, hidroklorotiazid memiliki waktu retensi sekitar menit ke-7, furosemid sekitar menit ke-12, dan spironolakton sekitar menit ke-15.
Setelah kondisi optimum KCKT untuk pemisahan hidroklorotiazid, furosemid, dan spironolakton didapatkan, kemudian dilakukan validasi. Pertama dilakukan penentuan linieritas dengan penyuntikan larutan baku hidroklorotiazid, furosemid, dan spironolakton. Pengukuran yang dilakukan menghasilkan persamaan garis linier dengan koefisien korelasi di atas 0,990. Pembuktian adanya hubungan linier antara konsentrasi analit dengan respon
instrumen dilakukan melalui uji linieritas. Penilaian dapat ditentukan dengan kriteria koefisien variasi fungsi regresi (Vx0) dan koefisien korelasi (r). Syarat (Vx0) untuk analisis obat dalam matriks biologis lebih kecil atau sama dengan 5%, sedangkan nilai r2 lebih besar atau sama dengan 0,99 (Ibrahim, 2005).
Batas deteksi (BD) dan batas kuantisasi (BK) dihitung dengan metode Miller dan ditentukan dari data linieritas. Penentuan BD dan BK dihitung dari kurva baku yang diajukan oleh Miller (Ibrahim, 2004). Batas deteksi didefinisikan sebagai konsentrasi analit terkecil yang memberi sinyal instrumen yang berbeda secara nyata dari sinyal blangko dan sinyal latar belakang, sedangkan batas kuantisasi didefinisikan sebagai konsentrasi analit terkecil yang dapat dikuantisasi secara cermat dan seksama.
Hasil penentuan linieritas koefisien korelasi, batas deteksi dan batas kuantisasi dapat dilihat pada tabel 3.
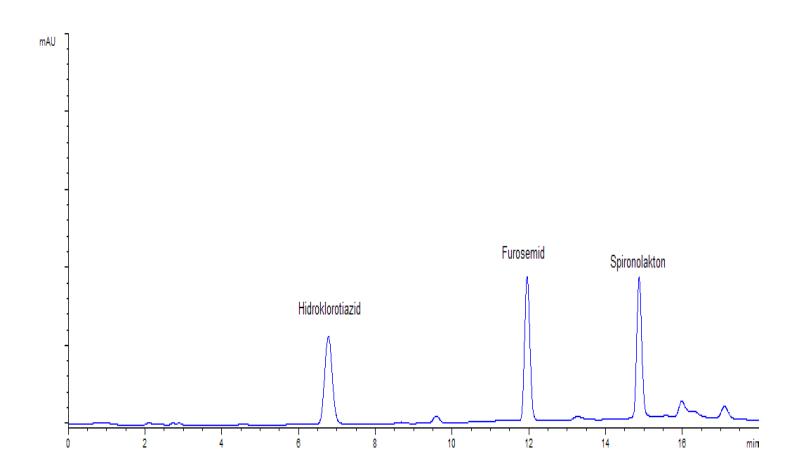
Gambar 2. Kromatogram larutan baku hidroklorotiazid (4 ppm), furosemid (4 ppm), dan spironolakton (10 ppm)
Kecermatan adalah ukuran yang menunjukan kedekatan hasil analisis dengan kadar analit yang dinyatakan dengan persen perolehan kembali (% recovery). Pengujian dilakukan dengan spikedplacebo recovery method, yaitu dengan menambahkan analit pada matriks yang diketahui konsentrasinya. Hasil perolehan kembali hidroklorotiazid, furosemid, dan spironolakton pada urin menunjukan hasil yang baik. Hasil lengkap dari uji perolehan kembali dapat dilihat pada tabel 4.
Untuk memvalidasi kekuatan suatu metode perlu dibuat perubahan metodologi kecil yang terus menerus dan mengevaluasi respons analitik. Identifikasi kekuatan metode dilakukan sekurang-kurangnya terhadap 3 faktor analisis yang dapat mempengaruhi
hasil bila diganti atau diubah (Harmita, 2004). Ketegaran (robustness) didefiniskan sebagai ukuran kemampuan metode untuk tetap tak berpengaruh dan bertahan terhadap pengaruh kecil, tapi dilakukan secara sengaja dengan membuat variasi dalam faktor metode yang memberikan indikasi reliabilitas metode normal pada pengujian.
Uji ketegaran dilakukan dengan merubah fase alir (normal 1 mL/menit) menjadi 0,9 mL/menit (-) dan 1,1 mL/menit (+), merubah pH dapar fosfat (normal pH 3) menjadi pH 2,8 (-) dan 3,2 (+), serta merubah panjang gelombang (normal 229 nm) menjadi 227 nm (-) dan 231 nm (+). Hasil pengujian ketegaran metode dan pengolahan datanya dengan ANOVA dapat dilihat pada tabel 5 dan tabel 6.
Tabel 3. Hasil penentuan linieritas, batas deteksi dan batas kuantisasi
| Analit | Persamaan garis | Koef. Korelasi (r) | *) Sy/x | **) Vxo | BD | BK |
|---|---|---|---|---|---|---|
| HCT | Y=136,88x + 1,046 | 0,9997 | 14,01 | 1,46% | 0,3 ppm | 1,0 ppm |
| Fur | Y=147,72x - 52,761 | 0,9991 | 26,60 | 2,57% | 0,5 ppm | 1,8 ppm |
| Spi | Y=60,006x + 4,876 | 0,9998 | 11,23 | 1,07% | 0,5 ppm | 1,9 ppm |
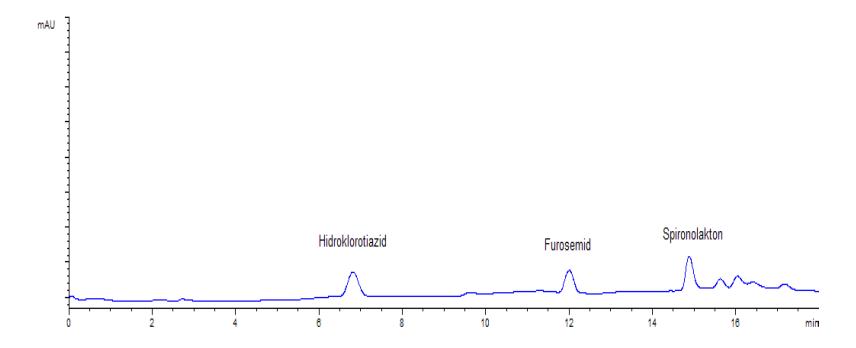
Gambar 3 : Kromatogram urin blangko
Tabel 4. Perolehan Kembali, Akurasi, dan Presisi Metode
| Analit | Konsetrasi (ppm) | % Recovery Absolut | %KV |
|---|---|---|---|
| Hidrokorotizid | 0,5 | 98,71 ± 0,24 | 0,25 |
| 5,0 | 98,57 ± 0,05 | 0,05 | |
| 10,0 | 99,08 ± 0,32 | 0,32 | |
| Rata-rata | 98,79 ± 0,27 | 0,27 | |
| Furosemid | 0,5 | 99,14 ± 0,27 | 0,28 |
| 5,0 | 98,97 ± 0,14 | 0,14 | |
| 10,0 | 98,99 ± 0,03 | 0,03 | |
| Rata-rata | 99,03 ± 0,09 | 0,10 | |
| Spironolakton | 1,0 | 99,05 ± 0,48 | 0,48 |
| 10,0 | 98,72 ± 0,05 | 0,05 | |
| 20,0 | 97,90 ± 1,26 | 1,29 | |
| Rata-rata | 98,56 ± 0,59 | 0,60 |
Tabel 5. Hasil perolehan kembali (%) pengujian ketegaran metode
| Analit | Laju alir | pH | Panjang gelombang | ||||
|---|---|---|---|---|---|---|---|
| (-) | (+) | (-) | (+) | (-) | (+) | ||
| Hidroklorotiazid 97,86 | 97,80 | 98,29 | 98,43 | 124,49 | 79,05 | ||
| Furosemid | 98,33 | 98,83 | 98,87 | 98,30 | 98,49 | 99,13 | |
| Spironolakton | 99,30 | 98,49 | 98,26 | 98,82 | 88,84 | 109,90 | |
Tabel 6. Hasil pengolahan ketegaran metode dengan ANOVA (α=0,05)
| Analit | FHitung | FTabel | Keputusan*) | Keterangan | ||
|---|---|---|---|---|---|---|
| Hidroklorotiazid | ||||||
| Pengaruh laju alir 2,241 | 9,30 | Ho | Diterima Tidak berpengaruh | |||
| Pengaruh pH | 1,825 | 9,30 | Ho Diterima | Tidak berpengaruh | ||
| Pengaruh | 108763,4 | 9,30 | Ho Ditolak | Berpengaruh | ||
| Furosemid | ||||||
| Pengaruh laju alir 0,289 | 9,30 | Ho Diterima Tidak berpengaruh | ||||
| Pengaruh pH | 2,124 | 9,30 | Ho Diterima | Tidak berpengaruh | ||
| Pengaruh | 7,674 | 9,30 | Ho Diterima | Tidak berpengaruh | ||
| Spironolakton | ||||||
| Pengaruh laju alir 1,242 | 9,30 | Ho | Diterima | Tidak berpengaruh | ||
| Pengaruh pH | 5,733 | 9,30 | Ho Diterima | Tidak berpengaruh | ||
| Pengaruh | 1118,1 | 9,30 | Ho Ditolak | Berpengaruh | ||
*) Ho : Tidak terdapat perbedaan akibat perubahan perlakukan.
Uji ketegaran menunjukan bahwa perubahan kecil pada laju alir dan pH tidak berpengaruh signifikan terhadap perolehan kembali hidroklorotiazid, furosemid dan spironolakton, sedangkan perubahan panjang gelombang memberikan pengaruh signifikan terhadap perolehan kembali hidroklorotiazid dan spironolakton tetapi tidak terhadap furosemid. Perubahan panjang gelombang terhadap perolehan kembali hidroklorotiazid dan spironolakton didukung oleh data spektrofotometer uv-vis bahwa penurunan atau penaikan kecil panjang gelombang pada detektor KCKT akan memberikan pengaruh terhadap serapan kedua analit tersebut.
Kesimpulan
Telah dikembangkan metode kromatografi cari kinerja tinggi (KCKT) yang tervalidasi untuk analisis hidroklorotiazid, furosemid, dan spironolakton dalam urin. Metode analisis senyawa-senyawa tersebut dilakukan menggunakan kolom LichroCART C18 (4,0 mm, 250 mm, 10 m) dengan sistem elusi gradien menggunakan asetonitril-dapar fosfat pH 3 sebagai fase gerak, laju alir 1,0 mL/menit dan menggunakan detektor ultraviolet pada panjang gelombang 229 nm. Hasil validasi metode menunjukan linieritas yang baik dengan koefisien korelasi (r) > 0,998 untuk hidroklorotiazid furosemid dan spironolakton. Koefisien variasi fungsi regresi (Vx0) untuk hidroklorotiazid adalah 1,46%, untuk furosemid 2,57%, dan untuk spironolakton 1,07%. Batas deteksi untuk hidroklorotiazid, furosemid, dan spironolakton berturut-turut adalah 0,3 ppm, 0,5 ppm dan 0,5 ppm, sedangkan batas kuantisasi untuk hidroklorotiazid, furosemid, dan spironolakton berturut-turut adalah 1,0 ppm, 1,8 ppm dan 1,9 ppm. Perolehan kembali untuk hidroklorotiazid, furosemid, dan spironolakton berturut-turut adalah 98,79%, 99,03% dan 98,56%. Uji keseksamaan memberikan nilai koefisien variasi sebesar 0,55% untuk hidroklorotiazid, untuk furosemid 0,44% dan untuk spironolakton 0,95%. Uji ketegaran menunjukan bahwa perubahan kecil pada laju alir dan pH tidak berpengaruh signifikan terhadap perolehan kembali hidroklorotiazid, furosemid dan spironolakton, sedangkan perubahan panjang gelombang memberikan pengaruh signifikan terhadap perolehan kembali hidroklorotiazid dan spironolakton tetapi tidak terhadap furosemid. Berdasarkan hasil pengujian secara keseluruhan dapat disimpulkan bahwa metode simultan penentuan hidroklorotiazid, furosemid, dan spironolakton secara KCKT telah berhasil didapatkan serta mampu memenuhi kriteria validasi metode analisis.
Saran
Metode KCKT yang telah dilakukan dapat dicoba diterapkan untuk mendeteksi penyalahgunaan hidroklorotiazid, furosemid, dan spironolakton sebagai doping oleh atlet olahraga terutama pada turnamen yang membutuhkan sertifikasi WADA atau IOC.
Perlu dilakukan pengembangan metode KCKT terhadap senyawa-senyawa diuretik menggunakan kombinasi fase gerak selain asetonitril karena tepat setelah berakhirnya penelitian ini, asetoniril menjadi sulit didapat, walaupun tersedia harganya sangat mahal.